
DOI�� 10.11817/j.issn.1672-7207.2020.12.004
�����ܶȷ������۵�KF-NaF-AlF3���µ������ϵ�Ʋ�����ϼ����Ӷ���ѧģ��
������1, 2������1���ź���1, 3�����1, 3
(1. ���ϴ�ѧ ұ���뻷��ѧԺ������ ��ɳ��410083��
2. �й����ż�������˾��ɽ�� ��ƽ��256200��
3. ���ϴ�ѧ ��ұ��ɫ������Դ��Ч���ù��ҹ���ʵ���ң����� ��ɳ��410083)
ժҪ�������ܶȷ������۹���KF-NaF-AlF3������ϵ��Buckingham�Ʋ������������������Ʋ�����KF-NaF-AlF3������ϵ���з��Ӷ���ѧģ�⡣�о������������������Ҫ�������ӹ���ΪF-����AlF4��-����AlF5��2-�ͣ�AlF6��3-�������ŷ����ӵĴ��ڣ��������γ��˸����ӵ�Al-F-Al������ӹ��ͣ����Al2F8��2-����Al3F10��-����Al4F14��2-����Al4F15��3-�ͣ�Al4F16��4-�ȣ���Щ���������ӵĴ��ڿ��ܻ��һ������KF-NaF-AlF3���ε��������ܡ�
�ؼ��ʣ��Ʋ�����ϣ�KF-NaF-AlF3�����Ӷ���ѧģ�⣻���ӽṹ
��ͼ����ţ�TF821 ���ױ�־�룺A ���ſ�ѧ(��Դ����)��ʶ��(OSID)
���±�ţ�1672-7207��2020��12-3300-09
Potential parameter fitting and molecular dynamics simulation of KF-NaF-AlF3 low temperature electrolyte system based on density functional theory
WANG��Jingkun1, 2, GUO��Hui1, ZHANG��Hongliang1, 3, LI��Jie1, 3
(1. School of Metallurgy and Environment, Central South University, Changsha 410083, China;
2. China Hongqiao Group Limited, Zouping 256200, China;
3. National Engineering Laboratory for High Efficiency Recovery of Refractory Nonferrous Metals,
Central South University, Changsha 410083, China)
Abstract: Buckingham potential parameters of KF-NaF-AlF3 molten salt system were constructed based on density functional theory.Molecular dynamics simulation of KF-NaF-AlF3 molten salt system was carried out based on the constructed potential parameters. The results show that the main anion configurations in the molten salt are F-, [AlF4]-,[AlF5]2- and [AlF6]3-. Due to the presence of bridge F ions, more complex Al-F-Al complex ion configurations are formed in the molten salt, such as [Al2F8]2-, [Al3F10]-, [Al4F14]2-, [Al4F15]3- and [Al4F16]4-, etc. The existence of these complex anions may further reduce the transport performance of the molten salt KF-NaF-AlF3.
Key words: potential parameter fitting; KF-NaF-AlF3; molecular dynamics simulation; ionic structure
�����������һ���ܺĽϸߵĹ��գ�����1 t Al��������ĵ�12 000~14 000 kW��h(�����ɱ�ռ������ܳɱ���35%~40%)��100����������������õĵ���ʼ���û�б仯��һֱ��NaF-AlF3��ϵΪ��[1]��NaF-AlF3��ϵ�ij����¶Ƚϸߣ�һ��Ϊ910~960 ��(NaF�����ʵ�����AlF3�����ʵ���֮��Ϊ2.0~2.7)���⼫��������˵�����������ɱ�[2]�����͵������ϵ�ij����¶ȿ�����Ч�����ܺģ���ˣ�Ѱ��һ�־��нϵͳ����¶Ⱥ��ȶ�������ѧ���ʵ����͵������һ�ֽϺõĽ���취��KF-AlF3���������ϵ���б�NaF-AlF3��ϵ���͵ij����¶�(����800 ��)���Ҷ����������ܽ�������ǿ���õ����о����ǵĹ㷺��ע[3-5]�������ڵ��������ƵĻ���(��������ԭ����������������)��KF-AlF3�������ϵ����ת��ΪKF-NaF-AlF3�������ϵ����ˣ�KF-NaF-AlF3������ϵ�ǽ����˵ĵ������������ϵ��Ŀǰ�����Ƕ�KF-NaF-AlF3��ϵ��ʵ���о����٣������о���Ҫ������һЩ������������ѧ������������ܶȺ�Һ�����¶ȵ�[1-3]������KF-NaF-AlF3��ϵ�����ӽṹ�о����١���չ���������ʵ���������ѵ㣺1) ���������ϵ�ĸ���ǿ��ʴ�Զ�ʵ���豸����Ľϴ�2) ���ڷ������ӷ�������ȷ�ⶨ��ṹ�����ʣ�3) ���ڷ�����Ľṹ�ϸ��ӣ�ʵ���о����ѡ�������������[6]���˴Ź���[7]�Լ������ģ�⼼���ķ�չ[8-12]��������ʸ���������о�ȡ��������ɹ�����������������Լ��˴Ź������������ģ�⼼�������ǵ�һ��ԭ���ͷ��Ӷ���ѧģ�⼼�����кܴ�ijɱ�����[13-18]����ֱ�Ӵӵ�һ��ԭ�����������������������ϵ������ȫ���ӻ�ѧ�����������ڼ��㷽���ϴ���һ�����ѣ�������ʱ�����ƣ�����ȫ��ؽ��м����ģ�⡣���������о����dz��Խ�ϵ�һ��ԭ���;���ķ��Ӷ���ѧģ�������һ��ԭ�����Ӷ���ѧģ��(FPMD)��������KCl-LiCl[13]��Li2BeF4[14]��LiF-NaF-KF[15]����ϵ�õ��˺ܺõ�Ӧ�á�FPMD����������������ѡ���ƺ������Ʋ���������ֱ�ӻ��ڵ�һ��ԭ�����㡣�����ַ����ܼ����ԭ�Ӹ������٣�һ��ֻ��100~200��ԭ�ӣ�������ʵ��ϵ�нϴ��࣬����ģ��ʱ��϶̣���Ϊ10 ps����[13]������FPMD�����Ѿ��������죬������KF-NaF-AlF3������ϵ��FPMD�о������١�LU��[19]����FPMD������KF-NaF-AlF3��ϵ�����ӽṹ���������ʽ�����ģ�⣬��������ϵ�����������٣�ģ��ʱ��϶̣�����ȫ��ӳKF-NaF-AlF3��ϵ��ʵ�����ӽṹ��ʽ������LU��[19]���о���ϵ�볣��ĵ��µ������ϵ����ɲ�ͬ[3]����ͬ��ɵĵ������ϵ�����ʴ���һ�������һ��������ģ�ͻ����ϣ����÷��Ӷ���ѧģ�ⷽ���о������������Ľṹ��������һ�ֽϺõ��ֶ�[20]����չKF-NaF-AlF3��ϵ�ķ��Ӷ���ѧģ����ؼ��ľ����ƺ����Լ�����Ʋ�����ѡ��Buckingham����Ϊһ�ֶ������Ѿ��㷺Ӧ����NaF-AlF3��ϵģ��[17]����KF-NaF-AlF3��ϵ��Ӧ���Ʋ������٣���������÷��Ӷ���ѧģ�ⷽ������һ�����ѣ�Ϊ�ˣ������������Ȼ���DFT���ۼ��㹹��KF-NaF-AlF3��ϵ��Buckingham�Ʋ�����Ȼ���ڹ������Ʋ������þ�����Ӷ���ѧģ���о�KF-NaF-AlF3��ϵ�����ӽṹ����仯���ɣ��Ա�Ϊ���µ���ʵ�ʵ���о��ṩ����֧�š�
1 ���۷���
1.1���ƺ���ģ��
KF-NaF-AlF3��ϵ��ÿ�����Ӷ��ھ���Ķ���������Ҫ����3���������[21]����ϵ�в�ͬ���ӶԼ�Ŀ����������ľ����������Vcharge����ϵ�����ӵĵ������ص������ij����������Vrepulsion��ɫɢ��������ɫɢ�������Vdispersion�����������Vtotale=Vcharge+Vrepulsion+Vdispersion��ɫɢ��������ɫɢ�������Vdispersion����ϵ�����ӵ��������˲�䲻�غ����£�����ֻ����ɫɢ���е�ż��-ż������á����IJ���Buckingham�ƺ���������ϵ�ij�������ú�ż��-ż�������[17]����ϵ��Buckingham���Լ�����������Ƶĺ�����ʽ���£�
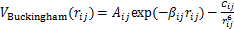 (1)
(1)
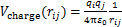 (2)
(2)
ʽ�У� Ϊ2������֮����룻
Ϊ2������֮����룻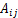 ��
�� ��
��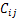 Ϊ��������������֮�������õIJ�����
Ϊ��������������֮�������õIJ����� Ϊ
Ϊ ����������ɣ�
����������ɣ� Ϊ��ս�糣����
Ϊ��ս�糣����
1.2���Ʋ����Ĺ���
�ڹ���������ķ��Ӷ���ѧģ������У��о�����Թ����εĻ����ṹ��Ԫ�����������壬�����һ��ԭ���ĵ����ܽ��м��㣬ͨ������������ԭ�Ӽ����ı仯����ԭ�Ӽ��������ƣ������ø����������Ӧ���Ʋ���[20]�����ǵ������������ϵͬ�����ڸ�����ϵ����������ṹ���Ӷ�䣬������������������Ʋ�����Ϸ�������KF-NaF-AlF3��ϵ�в�ͬ���ӶԵ�һ��ԭ�������ܽ��м��㣬�õ�Al-Al��Al-F��F-F��K-Al��K-F��K-K��K-Na��Na-Al��Na-F��Na-Na���ӶԵ��������ߣ������ܽ��зֽ�õ�Buckingham���ܲ�������ϡ�
1.2.1����ͬ���ӶԵ����ܵļ��㡡
���ù����ݶȽ���(GGA)�е�Perdew-Burke-Ernzerhof(PBE)���������[18]�����һ��ԭ�����ӶԵĵ����ܡ�ʹ��Ultrasoft������������������ʵ�ͼ۵��ӵ�����ã����У�����Na 2s22p63s1��Al 3s23p1��K 3s23p64s1��F 2s22p5����Ϊ�۵��ӡ�DFT-D2����[12]���ڴ������Ӽ�ķ��»�������á��Լ������õĽض��ܡ�K�������ģ�ͺ��ӵ������Խ��ж�β��ԣ����ýض���Ϊ850 eV��10��10��1��K������ֲ��ͳ���������Ϊ(15��10-10) m��(15��10-10) m��(15��10-10) m�ĺ��ӣ����м������CASTEP[22]ģ������ɡ����ݼ���õ��IJ�ͬ���ӶԵĵ����ܣ��������������Ӽ����仯��������ƣ����ں��������ֽܷ⡣��˵�����ǣ��ڻ����ܶȷ������۵ĵ�һ��ԭ�������У�����������������Ӽ����仯���仯��������ģ����������������ǹ̶�����ģ���ˣ��б�Ҫ��KF-NaF-AlF3��ϵ�в�ͬ����������ɽ����о���
1.2.2��KF-NaF-AlF3��ϵ�����ӵ�ɵ�ȷ����
����ʽ(2)��֪���ڶԾ�����������ܽ��зֽ�ʱ������Ҫȷ������������ɡ����е�һ��ԭ������ʱ������������������仯�����ϱ仯��������ķ���ģ�������Ϊ����ǹ̶�����ġ�Ϊ�˽����һ���⣬��������õ�һ��ԭ��ģ����ʵ��KF-NaF-AlF3������ϵ��ͨ������KF-NaF-AlF3��ϵ�в�ͬ���ӵ�ƽ��Mulliken���ȷ�����յķ���ģ��������������ɡ�����FPMD�����KF-NaF-AlF3��ϵ�Ļ�ѧ���Ϊ11��K���ӡ�9��Na���ӡ�15��Al���Ӻ�65��F���ӣ���100�����ӡ����ù̶���������������¶ȵ�NVTϵ�۽���FPMDģ�⣬�¶�����Ϊ827 �棬�Ը���KF-NaF-AlF3������ϵ�ij����¶�[23]��ģ����ӵ��ܶ�[3]����Ϊ1.846 g/cm3��������ϵ����NVTϵ���½���6 000��ģ�⣬�õ��ӽ���ʵ��ϵ���������ӽṹ��Ȼ�����4 000���ṹ��ԥ��ģ��ʱ�䲽������Ϊ1 fs���ܵ�ģ��ʱ��Ϊ10 ps����������������1.2.1���е���ͬ������FPMD����ģ��õ����յ��ȶ��������ڷ���KF-NaF-AlF3��ϵ������������ɡ�
��KF-NaF-AlF3��ϵ�У�����ע����Al-F���Ӽ������ã���Ϊ�������ӶԵ�����ý����������γ��ȶ��Ļ�����[24]��������һԭ�������ؿ���KF-NaF-AlF3��ϵ�з�����������ɣ�����������������ɸ��ݵ�����ԭ��õ���ͨ����FPMD����õ���KF-NaF-AlF3��ϵ���ȶ����ͽ���Mulliken��ɷ������õ���ϵ��65��F����������ɣ���ͼ1��ʾ�����������������ӵ����-0.71 e��-0.63 e֮�䲨��(���У�eΪ����)��ƽ��ֵԼΪ-0.67 e����ˣ����Ľ�KF-NaF-AlF3��ϵ��F���ӵĵ����Ϊ-0.67 e�����ݵ�����ԭ��Na��K��Al���ӵĵ��������Ϊ+0.67 e��+0.67 e��+2.01 e��

ͼ1��KF-NaF-AlF3��ϵ�з����ӵ�Mulliken���
Fig. 1��Mulliken charge of F ions in KF-NaF-AlF3 system
1.2.3��������������ֽܷ⡡
KF-NaF-AlF3��ϵ�в�ͬ���ӶԵ�Buckingham���ܿ��Ը��������ֽܷ�õ�������1.1�ڵķ�����Buckingham���ܵļ���ʽ���£�
 (3)
(3)
����1.2.2�ڵ�KF-NaF-AlF3��ϵ�����ӵ�ɵļ����������Եõ���ͬ���ӶԵľ���������ƣ�Ȼ��������ܽ��зֽ�õ���ͬ���ӶԵ�Buckingham���ܡ�Al-F�����ӵĽṹ������KF-NaF-AlF3��ϵ�����ʾ�����ҪӰ��[24]��ͼ2��ʾΪAl-F���ӶԵ��������ߡ���ͼ2�ɼ���Al-F���ӶԵ�������������ı仯��1.5��10-10~2.0��10-10 m֮��������Ե���͵㣬������KF-NaF-AlF3��ϵ����Ҫ����������������ǿ����õ����֣���ΪAl-F���Ӽ�ļ������ϴ������ǿ�������������γ��ȶ�����λ�ṹ��������һ������͵�����Ӧ�ľ�����������Al-F���ӵ�ƽ���������Ӧ[18-19]����˵�����Ĺ������Ӷ�������ļ�������Ǻ����ġ��������������ӶԵ�����������ͼ3��ʾ����ͼ3�ɼ�����Na-F���Ӷ��⣬��δ�������Ե��������ߵ���͵㡣��˵���������ӶԵļ��Բ����С�������������

ͼ2��Al-F���ӶԵ���������
Fig. 2��Potential energy curves of Al-F ion pairs

ͼ3��KF-NaF-AlF3��ϵ�в�ͬ���ӶԵ���������
Fig. 3��Potential energy curves of different ion pairs in KF-NaF-AlF3 system
1.2.4���Ʋ�����ϡ�
������С����ԭ����1.2.3�ڵõ���Buckingham�������߽��з�����������ϣ�������Ϲ��̾���Matlabƽ̨����ɣ�ԭ�����£�
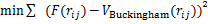 (4)
(4)
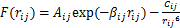 (5)
(5)
 (6)
(6)
ʽ(4)Ϊ��С���˷����з�������ϵĻ���ԭ�������У�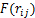 Ϊ��ϵ��ƺ�����
Ϊ��ϵ��ƺ�����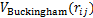 Ϊ��������ƺ�����ʽ(5)ΪBuckingham�ƺ��������в��������DZ�����Ҫ��ϵ��Ʋ���������1.1�ڵķ�����֪�����IJ���Buckingham�ƺ���������KF-NaF-AlF3��ϵ�в�ͬ���Ӷ�֮��ķǾ���������ƣ�ʽ(6)��ʾ��ϵ�еķǾ���������ơ���ϵõ����յ�Buckingham�Ʋ������1��ʾ��
Ϊ��������ƺ�����ʽ(5)ΪBuckingham�ƺ��������в��������DZ�����Ҫ��ϵ��Ʋ���������1.1�ڵķ�����֪�����IJ���Buckingham�ƺ���������KF-NaF-AlF3��ϵ�в�ͬ���Ӷ�֮��ķǾ���������ƣ�ʽ(6)��ʾ��ϵ�еķǾ���������ơ���ϵõ����յ�Buckingham�Ʋ������1��ʾ��
��1��KF-NaF-AlF3��ϵ�в�ͬ���ӶԵ�Buckingham�Ʋ���
Table 1��Buckingham potential parameters of different ion pairs in KF-NaF-AlF3 system

1.3�����Ӷ���ѧģ��
�����о���KF-NaF-AlF3������ϵ��ѧ��ɺ�ģ����ӵ�������2��ʾ����ʼ�ṹģ����Packmol����[25]����ؽ�����Ͷ�ŵ�ָ����С�ĺ����еõ����ھ�����Ӷ���ѧģ���У�ʹ��Verlet�����㷨���ţ���˶����̣�ʱ�䲽������Ϊ1 fs��EWALD����㷨[18]��Ϊ�������������Ӧ�����ձ�ķ����������ﱻ���ڴ�����ϵ�����Ӽ�Ŀ�������ú�ż������ã�ͬʱ��Buffer��������Ϊ0.5��10-10 m���������㾫��Ϊ4.184��10-5 kJ/mol[19]���̳�����õĽضϰ뾶Ϊ15��10-10 m�����ӵĵ������Ϊ+0.67 e(Na)��+2.01 e(Al)��-0.67 e(F)��+0.67 e(K)������Դ��1.2.2�ڵ�DFT�������������Ա߽�����������ģ����ӣ���������1��û�б߽������Һ̬ϵͳ��ʹ�ü��������ӿɿ���ģ�����ϵͳ������NPT[24]ϵ������0.1 MPaѹ�����µ�827 �棬ģ��ʱ��Ϊ2 000 ps������ζ��ģ������б���ϵͳ��������(N)��ѹ��(P)���¶�(T)Ϊ������֮��ʹ��NVTϵ�ۼ�������3 000 ps�ṹ��ԥ�����ʹ��Matlab����ͳ�Ʒ����ṹ��ԥ3 000 ps��ģ������е����ӹ켣���ݣ�����KF-NaF-AlF3���ε����ӽṹ�����еķ��Ӷ���ѧ�������Forciteģ�������[26]��
��2�����Ӷ���ѧģ������
Table 2��Molecular dynamics simulation conditions

2 ���������
2.1��KF-NaF-AlF3������ϵ�ľֲ����ӽṹ
���¶�Ϊ827 �桢ѹ��Ϊ0.1 MPa��ģ��õ�KF-NaF-AlF3������ϵ���ȶ�������ͼ4��ʾ����ͼ4���Կ�����K��Na��������ֲ���ģ������У���������֮��ľ���ϴ�������ΪK-K��K-Na��Na-Na�����Ӽ��Բ����С���Ҿ��������ԡ�ģ������о�������ӽṹ��������λ[AlF4]-������λ[AlF5]2-������λ[AlF6]3-������Ӽ����������ֱ��ӦŤ���������塢����˫��Ͱ����幹�͡�ͬʱ�������ŷ����Ӵ��ڣ��������γ���Al-F-Al�������ӹ��͡�KF-NaF-AlF3������ϵ��Ȼʧȥ�˳�������״̬�����������ӽṹ��Ȼ�����Ŷ̳����������д��ڴ���������λ[AlF4]-������λ[AlF5]2-������λ[AlF6]3-������Ӽ��š�

ͼ4��KF-NaF-AlF3������ϵ�������ӽṹ (KF��������Ϊ4%)
Fig. 4��Micro-structure of KF-NaF-AlF3 molten salt system
2.2������ֲ�����
����ֲ�����(RDF)��������Һ��ṹ���ɲ��ôӷ��Ӷ���ѧģ��켣��ȡ����RDF�������������εľ������ӽṹ[15]������ֲ�������ӳ���� ��������Ϊ���ģ��뾶
��������Ϊ���ģ��뾶 ��Χ�ڳ�����һ�����ӵĸ��ʡ�
��Χ�ڳ�����һ�����ӵĸ��ʡ�
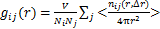 (7)
(7)
ʽ�У�VΪ���Ӷ���ѧģ����ӵ������NΪ������Ŀ�� Ϊ����
Ϊ���� ��ָ�������ضϷ�Χ��Χ������������ƽ����Ŀ��
��ָ�������ضϷ�Χ��Χ������������ƽ����Ŀ��
KF-NaF-AlF3��ϵ�����ӶԵ�RDF��ͼ5��ʾ����ͼ5�ɼ�����r>8��10-10 mʱ��g(r)��������1�����������������Զ������Ľṹ��ʽ���Ǻϣ�Al-F���ӶԵ�RDF��1.75��10-10 m���Ҵ���1���߶���ķ�ֵ������ζ��Al-F���Ӽ������ý�ǿ�������������γɽϸ��ӵ�������ӣ�������һ��ֵ��Al-F��ƽ���������Ӧ[18]��Al-Al���ӶԵ�RDF���Է�ӳ���������ӵľۺϳ̶ȣ���3.69��10-10 m��6.33��10-10 m���ֱ������Al-Al���ӶԵĵ�һ��ֵ�͵ڶ���ֵ����������[21]�����Ľ����һ�¡�Al-Al���ӶԵĵ�һ��ֵ�뾶ԼΪAl-F���ӶԵ�һ��ֵ�뾶��2������˵���������γ���Al-F-Al���͵ĸ���������ӣ�����������Ϊ����������2�������ӡ�һ�㾶��ֲ������ĵ�һ��ֵ������ijһ���ӶԵ�ƽ�����������������3��ʾ��
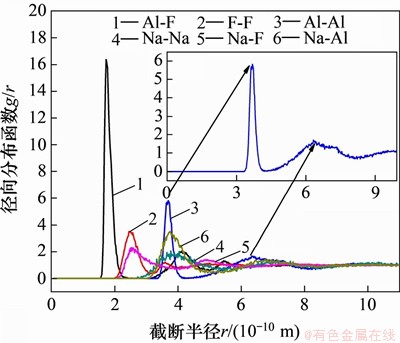
ͼ5��KF-NaF-AlF3��ϵ�����ӶԵľ���ֲ�����(RDF)(KF��������Ϊ4%)
Fig. 5��RDF of ion pairs in KF-NaF-AlF3 system (4% KF)
��3��KF-NaF-AlF3��ϵ�и����ӶԵ�ƽ������
Table 3��Average bond lengths of ion pairs in KF-NaF-AlF3 system 10-10 m

2.3�����Ƿֲ�
KF-NaF-AlF3������F-Al-F���Ƿֲ���ͼ6��ʾ�������[AlF6]3-������ӵ��8��90���3��180���F-Al-F���ǣ������[AlF5]2-����˫��ӵ��6��90�㣬3��120���Լ�1��180���F-Al-F���ǣ��������[AlF4]-����������ӵ��6��109.5���F-Al-F���ǡ���ͼ6���Կ�����F-Al-F�ļ��Ƿֲ����ߵĵ�һ��ֵ�͵ڶ���ֵ�ֱ�λ��91���116�㣬�ֱ��Ӧ��1�������������˫�������������Ӽ��Žṹ��ͬʱ��λ��173�����ҵĵ�����������˫���������������108�����ҷ����˵��ķ壬��Ӧ�������������Ӽ��Žṹ�������ֵ��С��˵��KF-NaF-AlF3�����д���������������λ[AlF4]-������Ӽ��Žṹ��ͬʱ����Щ���λ�ö���ƫ���������빹���б����ǵ�λ�ã�������Щ������Ӽ��Žṹ��������������Ť���ġ�ͨ�����������Ƿֲ�������з���������KF-NaF-AlF3������������Ӽ�����Ҫ������λ[AlF6]3-������λ[AlF5]2-Ϊ����ͬʱ��������������λ[AlF4]-������ӡ�

ͼ6��KF-NaF-AlF3������ϵ��F-Al-F���Ƿֲ� (KF��������Ϊ4%)
Fig. 6��F-Al-F bond angle distribution in KF-NaF-AlF3 system(4% KF)
2.4��F�������ͷֲ�
��ʵ�ϣ�KF-NaF-AlF3������Al-F�����ӵ����Ͳ����Ǽ�[AlF6]3-��AlF5]2-��[AlF4]-����Ϊ�����еķ����ӿ��ܻ���Ϊ����������2��Al�����γ�Al-F-Al���ͣ�����������������ӵĸ��ӳ̶ȡ�Ϊ�˽�����һ������ͳ�Ʒ�����KF-NaF-AlF3������ϵ�з��������ͷֲ����ɣ������ͼ7��ʾ��F��������Ϊ���ŷ�Fb����������Al-F-Al��ʽ����2�������ӣ��ն˷�Ft����������1���������������ӣ����ɷ�Ff��������û�����������������á�F�������;�����KF-NaF-AlF3�������ӽṹ�ľۺϳ̶ȣ��������εĴ������Բ���������Ҫ��Ӱ�졣��ͼ7���Կ���������KFŨ�������������ŷ����ӵ�Ũ���������ն˷����ӵ�Ũ����Ӧ���ͣ������ɷ����ӵ�Ũ��ά���ڽϵ�ֵ����˵������KFŨ������KF-NaF-AlF3������ϵ�����ӽṹ���ӣ���ϵ�����ӵ���ɢ����Ҳ����Ӧ���͡�

ͼ7��KF-NaF-AlF3������ϵ��F�������ͷֲ�
Fig. 7��F ion type distribution in KF-NaF-AlF3 system
2.5�������������ӽṹ���γ�
ͨ��ǰ�������֪��KF-NaF-AlF3������ϵ�д��ڳ����[AlF6]3-��[AlF5]2-��[AlF4]-����Щ��������ӹ��Ϳ��ܻ�ͨ���ŷ������γ������������ӵ������ӹ��͡�Ϊ�˸�ֱ�۵ر�ʾKF-NaF-AlF3������ϵ�и������ӽṹ���γɹ��̣�����ͳ������������Ҫ������Al-F������ӵĽṹ��ͨ��������Ӧ�ĽضϾ���(Al-F����ֲ������ĵ�һ��ֵ�뾶)[16]�����Լ���Al������ΧF���ӵ���Ŀ����ͼ8��ʾ����ͼ8��֪�������������ӹ�����Ҫ��F-��[AlF4]-��[AlF5]2-��[AlF6]3-��ͬʱ�������ŷ����ӵĴ��ڣ��������γ��˸����ӵ�������ӣ���[Al2F8]2-��[Al3F10]-��[Al4F14]2-��[Al4F15]3-��[Al4F16]4-�ȡ�

ͼ8��KF-NaF-AlF3������ϵ��Al-F������ӵĽṹ (KF��������Ϊ4%)
Fig. 8��Structure of Al-F complex ions in KF-NaF-AlF3 system (4% KF)
ͨ��ͳ�Ʒ���3 000 ps�����ӹ켣���ݣ��õ�KF-NaF-AlF3������ϵ�в�ͬAl-F������ӵİٷֱȣ����е�ͳ�ƽ��������1.3���еķ��Ӷ���ѧģ����̵õ�����ͼ9��ʾ������Al-F-Al��ʾ���ŷ��������ӵ�Al-F������ӹ��͡���ͼ9�ɼ�������KFŨ������������Al-F�����������Ը�����λ��[AlF6]3-���ڣ���[AlF5]2-��[AlF4]-�����ͣ���[AlF4]-һֱ��һ���������ͣ�����F-Al-F���Ƿֲ�������һ�¡����⣬Al-F-Al������ռ�ٷֱ����������������KFŨ���������ε����ӽṹҲ���ӣ������ӵ�Al-F�����ӹ��ͻή��Al���Ӻ�F���ӵ���ɢ���ܣ�������������������ϵ���������ܡ�

ͼ9��KF-NaF-AlF3������ϵ��Al-F����������ͷֲ�����
Fig. 9��Distribution of Al-F complex ion types in KF-NaF-AlF3 system
3 ����
1) ����DFT���۹�����KF-NaF-AlF3���µ������ϵ���Ʋ����������þ�����Ӷ���ѧģ���KF-NaF-AlF3��ϵ�������ӽṹ�������о���
2) KF-NaF-AlF3��ϵ����Ҫ��������ӹ���Ϊ[AlF4]-��[AlF5]2-��[AlF6]3-���ֱ��Ӧ��Ť���������塢����˫��Ͱ����幹�͡�KF-NaF-AlF3������ϵ��Ȼʧȥ�˳�������״̬�����������ӽṹ��Ȼ�����Ŷ̳�����Al-F���ӶԵ�RDF��1.75��10-10 m���Ҵ���1���߶���ķ�ֵ������ζ��Al-F���Ӽ������ý�ǿ�������������γɽϸ��ӵ�������ӣ�������һ��ֵ��Al-F��ƽ���������Ӧ��
3) F-Al-F�ļ��Ƿֲ����ߵĵ�һ��ֵ�͵ڶ���ֵ�ֱ�λ��91���116�㣬�ֱ��Ӧ��1�������������˫�������������Ӽ��Žṹ��ͬʱ��λ��173�����ҵĵ����巴ӳ������˫�����������108�����ҳ��ֵ��ķ壬��Ӧ�������������Ӽ��Žṹ�������ֵ��С����˵��KF-NaF-AlF3�����д���������������λ[AlF4]-������Ӽ��Žṹ��
4) �����ŷ����ӵĴ��ڣ��������γ��˸����ӵ�������ӣ���[Al2F8]2-��[Al3F10]-��[Al4F14]2-��[Al4F15]3-��[Al4F16]4-�ȡ�����KFŨ������������Al-F�����������Ը�����λ��[AlF6]3-���ڣ���[AlF5]2-��[AlF4]-�����ʵ���֮�����ͣ���[AlF4]-һֱ��һ���������͡������ӵ�Al-F�����ӹ��ͻή��Al���Ӻ�F���ӵ���ɢ���ܣ�������������������ϵ���������ܡ�
�ο����ף�
[1] DANIELIK V, GABCOVA J. Phase diagram of the system NaF-KF-AlF3[J]. Journal of Thermal Analysis and Calorimetry, 2004, 76(3): 763-773.
[2] CASSAYRE L, PALAU P, CHAMELOT P, et al. Properties of low-temperature melting electrolytes for the aluminum electrolysis process: a review[J]. Journal of Chemical & Engineering Data, 2010, 55(11): 4549-4560.
[3] YAN Hengwei, YANG Jianhong, LI Wangxing. Surface tension and density in the KF-NaF-AlF3 based electrolyte[J]. Journal of Chemical & Engineering Data, 2011, 56(11): 4147-4151.
[4] ����, �պ�ά. KF-NaF-AlF3-X���µ������ϵ�о���չ[J]. �����, 2009(9): 28-30.
WANG Min, YAN Hengwei. Review on the KF-NaF-AlF3-X electrolyte system[J]. Light Metals, 2009(9): 28-30.
[5] ������, ������. NaF-AlF3-Al2O3-CaF2-LiF-MgF2-KFϵ��ҵ������ʳ����¶ȵ��о�[J]. �����, 2019(1): 34-39.
ZHANG Yanli, QIU Shilin. Research on the liquidus temperature of NaF-AlF3-Al2O3-CaF2-LiF-MgF2-KF industrial aluminum electrolyte[J]. Light Metals, 2019(1): 34-39.
[6] AKDENIZ Z, MADDEN P A. Raman spectra of ionic liquids: a simulation study of AlF3 and its mixtures with NaF[J]. The Journal of Physical Chemistry B, 2006, 110(13): 6683-6691.
[7] BESSADA C, RAKHMATULLIN A, ROLLET A L, et al. Lanthanide and actinide speciation in molten fluorides: a structural approach by NMR and EXAFS spectroscopies[J]. Journal of Nuclear Materials, 2007, 360(1): 43-48.
[8] NAM H O, BENGTSON A, VORTLER K, et al. First-principles molecular dynamics modeling of the molten fluoride salt with Cr solute[J]. Journal of Nuclear Materials, 2014, 449(1/2/3): 148-157.
[9] FRANK W B, FOSTER L M. The constitution of cryolite and NaF: AlF3 melts[J]. The Journal of Physical Chemistry, 1960, 64(1): 95-98.
[10] CRISTIGLIO V, HENNET L, CUELLO G J, et al. Ab-initio molecular dynamics simulations of the structure of liquid aluminates[J]. Journal of Non-Crystalline Solids, 2007, 353(18/19/20/21): 1789-1792.
[11] LACASSAGNE V, BESSADA C, FLORIAN P, et al. Structure of high-temperature NaF-AlF3-Al2O3 melts: a multinuclear NMR study[J]. The Journal of Physical Chemistry B, 2002, 106(8): 1862-1868.
[12] BENGTSON A, NAM H O, SAHA S, et al. First-principles molecular dynamics modeling of the LiCl-KCl molten salt system[J]. Computational Materials Science, 2014, 83: 362-370.
[13] BELONOSHKO A B, AHUJA R, JOHANSSON B. Molecular dynamics of LiF melting[J]. Physical Review B, 2000, 61(18): 11928.
[14] KLIX A, SUZUKI A, TERAI T. Study of tritium migration in liquid Li2BeF4 with ab initio molecular dynamics[J]. Fusion Engineering and Design, 2006, 81(1/2/3/4/5/6/7): 713-717.
[15] LU Xiaojun, HAN Zexun, CHEN Jiangan, et al. First-principles molecular dynamics study of ion structure and transport properties of LiF-NaF-AlF3 molten salt[J]. Chemical Physics Letters, 2018, 706: 237-242.
[16] MACHADO K, ZANGHI D, SAROU-KANIAN V, et al. Study of NaF-AlF3 melts by coupling molecular dynamics, density functional theory, and NMR measurements[J]. The Journal of Physical Chemistry C, 2017, 121(19): 10289-10297.
[17] LU Xiaojun, XU Zhenming, LI Jie, et al. Theoretical investigation on local structure and transport properties of NaF-AlF3 molten salts under electric field environment[J]. Journal of Molecular Structure, 2016, 1117: 105-112.
[18] LU Xiaojun, XU Zhenming, LI Jie, et al. First-principles molecular dynamics investigation on Na3AlF6 molten salt[J]. Journal of Fluorine Chemistry, 2016, 185(27): 42-47.
[19] LU Xiaojun, HAN Zexun, ZHANG Hengxing, et al. Ionic structure and transport properties of KF-NaF-AlF3 fused salt: a molecular dynamics study[J]. Physical Chemistry Chemical Physics, 2019, 21(14):7474-7482.
[20] TSUNEYUKI S, TSUKADA M, AOKI H, et al. First-principles interatomic potential of silica applied to molecular dynamics[J]. Physical Review Letters, 1988, 61(7): 869.
[21] MACHADO K, ZANGHI D, SALANNE M, et al. Anionic structure in molten cryolite-alumina systems[J]. The Journal of Physical Chemistry C, 2018, 122(38): 21807-21816.
[22] CLARK S J, SEGALL M D, PICKARD C J, et al. First principles methods using CASTEP[J]. Zeitschrift F��r Kristallographie-Crystalline Materials, 2005, 220(5/6): 567-570.
[23] APISAROV A, DEDYUKHIN A, REDKIN A, et al. Physical-chemical properties of the KF-NAF-ALF3 molten system with low cryolite ratio[C]// TMS Light Metals. Pennsylvania, USA, 2009: 401-403.
[24] LU Xiaojun, XU Zhenming, LI Jie, et al. Molecular dynamics investigation on structural and transport properties of Na3AlF6-Al2O3 molten salt[J]. Journal of Molecular Liquids, 2016, 221: 26-32.
[25] MARTINEZ L, ANDRADE R, BIRGIN E G, et al. PACKMOL: a package for building initial configurations for molecular dynamics simulations[J]. Journal of Computational Chemistry, 2009, 30(13): 2157-2164.
[26] BHUVANESHWARI B, PALANI G S, IYER N R. Multiscale analysis and mechanical properties of cement clinker phases with molecular dynamics simulations[J]. Journal of Coupled Systems and Multiscale Dynamics, 2014, 2(4): 214-223.
(�༭ �²ӻ�)
�ո����ڣ� 2020 -06 -16; �����ڣ� 2020 -08 -22
������Ŀ(Foundation item)��������Ȼ��ѧ����������Ŀ(51974373��51674300��51874365��61751312)������ʡ��Ȼ��ѧ����������Ŀ(2018JJ2521) (Projects(51974373, 51674300, 51874365, 61751312) supported by the National Natural Science Foundation of China; Project(2018JJ2521) supported by the Natural Science Foundation of Hunan Province)
ͨ�����ߣ���£���ʿ�����ڣ����������������ۼ������Ż������о���E-mail��13808488404@163.com

