���Ӷ���ѧģ��ͭ�����ŴصĽṹ�ȶ���
������1, 2, ������1, �� ��1, л ��1
(1. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410083;
2. ���մ�ѧ ���Ͽ�ѧ�빤��ѧԺ, �� 212013)
ժ Ҫ: ���÷��Ӷ���ѧ�о���ԭ����Ϊ13~1055��ͭ�����Ŵء� �������: ���ųߴ�ļ�С, ͭ�����ŴصĽṹ����������Ǿ���������Ǿ���������Ǿ���ת�䡣 �Ŵ�ƽ��ԭ�ӽ�������ųߴ�ļ�С����С, ��ֻ�����ڶ̳�����, ��˵�����Ŵ�ƽ��ԭ�ӽ����һ�㲻�ܹ���Ϊ�Ǿ����ܶѽṹ����ת����оݡ� ƽ��ԭ�Ӽ����������Ŵصijߴ�, �Ҷ��Ŵؽṹ�ı仯����, ������Ϊ�Ǿ����ܶѽṹ����ת���һ���оݡ� ��ż�ֲ��������о�����, ��ߴ��Ŵص��ڲ��ͱ���ԭ�ӽṹ�����ֳ���������ЧӦ, �Ҳ�ͬ����Ӧ���徧��, �������Ŀǰ�����й����ŴصĿ���ӱ���ģ����Ǻ�ģ�Ͷ��д��Ľ���
�ؼ���: ���Ӷ���ѧ; ͭ�Ŵ�; �����; ƽ��ԭ�Ӽ�� ��ͼ�����: O482.2; TG111.5
���ױ�ʶ��: A
Molecular dynamics simulating of structural stability of copper nanoclusters
QI Wei-hong1, 2, WANG Ming-pu1, LI Zhou1, XIE Dan1
(1. School of Materials Science and Engineering,Central South University, Changsha 410083, China;
2. School of Materials Science and Engineering,Jiangsu University, Zhenjiang 212013, China)
Abstract: The Cu nanoclusters with atom number of 13-1055 have been studied by molecular dynamics simulation method. The results show that the structure of Cu nanoclusters follows the sequence of crystalline��amorphous��crystalline��amorphous��crystalline��amorphous with the decrease of the cluster size. The average atom cohesive energy decreases with the decrease of the cluster size, and only depends on the short-range order, which shows that the mean atom cohesive energy can not be regarded as the criterion of determining the structure of amorphous and close packed crystallines. However, the mean atom distance depends on not only the cluster size, but also the cluster structure, which can be regarded as the criterion of determining the structure transformation. Furthermore, the study on pair correlation function shows that both the interior and outer shells of the larger clusters contract a little, and are different from those of the corresponding bulk crystal, which suggests that the present bulk-surface and core-shell models of clusters should be improved.
Key words: molecular dynamics; Cu nanoclusters; cohesive energy; mean atom distance
���������Ŵص��о��ǽ������һ���ȵ�, ��Ҫ�������Ŵر������Ƿ��Ӻͺ�۲��ϵĹ���̬[1,2], ��ܶ����ԼȲ�ͬ�ڵ�������, Ҳ��ͬ�ڿ���������¡� ���������Ŵ��ڴ��Լ��������������ŷdz��㷺����;[3-6]�� ���ϵ�����ȡ���ڽṹ, �����Ŵص�����Ҳ������������Ľṹ�� ���о���Ҫ����ͭ�����ŴصĽṹ���ԡ�
Taylor��[7]���ù�����о�������ͭ�����Ŵ�, Knickelbein[8]�ⶨ������ͭ�Ŵص��뻯�ܲ������˵��ӿDz�ṹ�� �������о�����, Massobrio��[9]�����ܶȷ�������������ԭ����nΪ2, 4, 6, 8��10��ͭ�Ŵؽṹ�Լ��������, Kabir��[10, 11]����FP-LMTO�����о���ԭ����С��9��ͭ�Ŵ�, �����÷��Ӷ���ѧ�о���nΪ10~55��ͭ�Ŵؽṹ���ԡ� �����������÷��Ӷ���ѧ�ṹ��ԥ�����о���ԭ����Ϊ13~1055ͭ�����Ŵص��ȶ��ṹ, ͨ������ͭ�Ŵؽ���ܺ�ƽ��ԭ�Ӽ�������̬��Ǿ�̬��һ���о�, ���������еĽ����Ŵص�ģ�͡�
1 ģ�ⷽ��
����Materials Explorer ���Ӷ���ѧ������ģ��, ���ý�������, ���ýṹ��ԥ�ķ���������Ŵ����ȶ��ṹ�� ��������ϵ��(NTV)ģ�⡣ �Ŵصij�ʼ��̬����ͨ������������Ϊ���������������ͭ������ȡ�����ε�ͭ�Ŵ�, ��ѡһ������ԭ��, �ӵ�1����һֱȡ����29����, �Ŵص�ԭ�����ֱ�Ϊ13�� 19�� 43�� 55�� 79�� 87�� 135�� 141�� 177�� 201�� 225�� 249�� 321�� 369�� 381�� 429�� 459�� 531�� 555�� 603�� 627�� 675�� 683�� 767�� 791�� 887�� 935�� 959��1055�� ��ÿ���Ŵطֱ���800K����5000���ﵽƽ��, Ȼ���¶ȴ�800K����0K, ������45000��, ����Ϊ2fs, �������ɱ߽�������
2 ���������
ͼ1��ʾΪͭ�ŴصĶ�ż�ֲ������� ��ͼ1�ɼ�, ��Ȼn=249ʱͭ�Ŵصij�ʼ�ṹΪ���뾧��, ��ͨ�����Ӷ���ѧ�ݻ�, �ܶ����ʧ, �����ȶ��ṹΪ�Ǿ�̬�� ����n=321��ͭ�Ŵ�, �Ӷ�ż�ֲ���������, ��ʼ�ṹ�Ǿ���, ͨ�����Ӷ���ѧ��ԥ��, ��λ������, ˵����ṹ���ֳ�������, ��Ȼ�Ǿ�̬�� ͨ���������ŴصĶ�ż�ֲ������о�����, ԭ����Ϊ19, 43, 55, 79, 87, 135, 141, 249��459���Ŵ�Ϊ�Ǿ�̬, ��ԭ����Ϊ177, 201, 225, 321, 369, 381, 429�Լ�n��531���Ŵ�Ϊ��̬�� ԭ����Ϊ13���Ŵ�ֻ��һ��ԭ��, ������Ϊ�Ǿ�̬, Ҳ������Ϊ�ǷǾ�̬�� ���Ӷ���ѧ��ԥ��Ϊ���ҳ��Ŵص���������� ���, �Ŵؽṹ�ʷǾ�̬, ˵���Ǿ�̬�������ܸ���, �ṹ���ȶ��� ���Ŵؽṹ����ԥ��ɾ�̬�Ŵ�, ˵���ṹΪ��̬, �����������͡� ���ֽṹ������ѡ����Ҫ�����������Ŵ��нϴ�ıȱ����, �ϴ�ıȱ��������ֱ��Ӱ���Ŵؽṹ���ܡ� ���ȱ�������Ӱ�������ŴصĽṹ�����һ�����о���
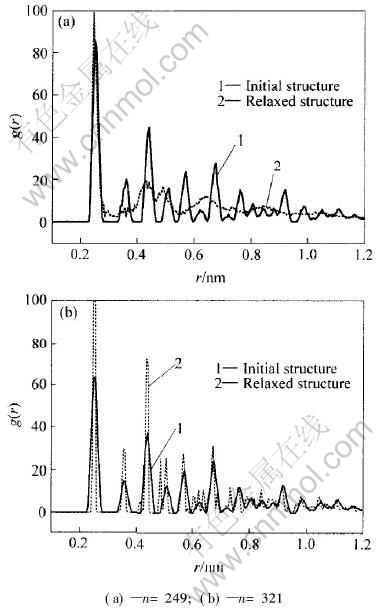
ͼ1 ͭ�ŴصĶ�ż�ֲ�����
Fig.1 Pair correlation function of Cu clusters
ͼ2��ʾΪͭ�����Ŵ�ƽ��ԭ�ӽ������ԭ�����Ĺ�ϵ�� ��ͼ2�ɿ���, �����Ŵص�ƽ��ԭ�ӽ���������Ŵسߴ�ļ�С�����͡� ��Ȼͭ�Ŵص��ȶ��ṹ�о�̬�ͷǾ�̬, ����ƽ��ԭ�ӽ����û�з���ͻ��, �������ųߴ�ı仯ƽ���ر仯�� ��˵���ŴصĽ���ܲ��������Ŵصij���������, ���������ڶ̳�������, ��̬�ͷǾ�̬�IJ�����ڳ�������, ���������ܴӽ���ܵIJ����ϱ��ֳ���; ƽ��ԭ�ӽ���ܲ�����Ϊ��̬��Ǿ�̬���оݡ�
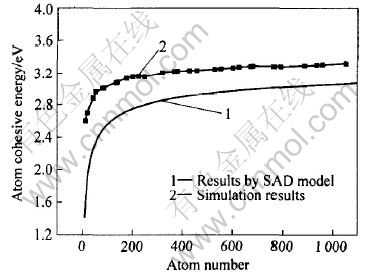
ͼ2 ͭ�ŴصĽ������ԭ�����Ĺ�ϵ[12, 13]
Fig.2 Relationship between cohesive energy and atom number of Cu nanoclusters[12, 13]
Ϊ�˱Ƚ�, ͼ2��Ҳ�����˱��������ģ��(surface area difference, SAD)����ͭ�Ŵ�ԭ�ӽ���ܵļ�����[12, 13]�� ��ͼ2�ɿ���, SADģ��Ԥ�����ͱ��о�ģ�����ı仯����һ�¡� SADģ�͵ĺ�������Ϊ: ������ǽ����Ϸֳɹ���ԭ������Ҫ������, �罫����ԭ��Ҳ����һ�����б��������ʵ��, ��ô, ���й���ԭ�ӵı�����ͽ�����ԭ���ϵı����, Ҳ����˵����ܵ�ֱ�ӽ���൱�ڲ������µı���, ����������Ͻ�, ����ܾ�Ӧ�õ������й���ԭ�ӵı���������ϱ�����֮� SADģ�͵ļ��㹫ʽΪ Ep=Eb(1-��n-1/3), ʽ��Ep��Eb�ֱ����ŴغͿ�����ϵĽ���ܡ� ����ͭ, Eb=3.49eV[14], ��Ϊ��״����; ���ڶ������������Ŵ�, ��=1.245[15]�� �ɴ˿ɿ���, SADģ��û�п��Ǿ����ԥ�� ���о��еķ��Ӷ���ѧģ����õ��DZ����ֳ�ԥ��Ľ����, ���ṹ��ԥ����ʹ�����ܽ���, �ṹ���ȶ�, ��Ȼ�����Ҳ���� ���, �ṹ��ԥ��ķ��Ӷ���ѧģ��Ľ���ܱ�δ��ԥ��SADģ�����Ľ����Ҫ��, ����ͼ2��ʾ�Ľ��һ�¡�
ͼ3��ʾΪͭ�Ŵ�ƽ��ԭ�Ӽ����ԭ�����Ĺ�ϵ�� ��ͼ3�ɿ���, ƽ��ԭ�Ӽ��Ҳ�������Ŵ�ԭ����, �Ŵص�ƽ��ԭ�Ӽ�С����Ӧ����ͭ��ƽ��ԭ�Ӽ�ࡣ �������ܲ�ͬ����, ƽ��ԭ�Ӽ���������Ŵؽṹ�� ���ھ��о�̬�ṹnΪ13, 177, 201, 225, 321, 369, 381, 429�Լ�n��531(����n=13�ĽṹҲ���ɾ�̬)���Ŵ�, ��ƽ��ԭ�Ӽ�������Ŵسߴ�������ƽ��������, �������ڿ���ͭ�����ƽ��ԭ�Ӽ�ࡣ �����ھ��зǾ�̬�ṹ��nΪ19, 43, 55, 79, 87, 135, 141, 249��459���Ŵ�, ��ƽ��ԭ�Ӽ������ִ�����ЧӦ, ʱ��ʱС, ���ܵ���˵, �Ǿ��Ŵص�ƽ��ԭ�Ӽ��Ҫ������Ӧ��̬��ƽ��ԭ�Ӽ�ࡣ �������̬ƽ��ԭ�Ӽ���ƽ���仯��ϳ�һ������, ��ô�Ϳ����ж������Ŵ��Ƿ���о�̬�ṹ�� ����õ���ƽ��ԭ�Ӽ������������, ���Ŵؾ�������ľ�̬�ṹ�� �����ƫ������, ����ṹҲ����Ӧ��ƫ�뾧̬, ��������Զ, ƫ��ij̶����� ������Ƕ���˵, �Ŵ�ƽ��ԭ�Ӽ�������Ϊ�ж��Ŵؽṹ��һ���оݡ�
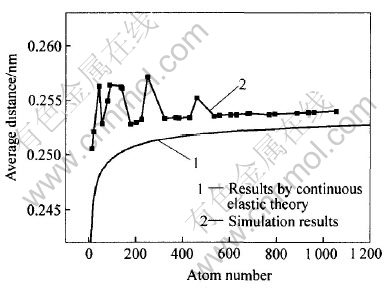
ͼ3 ͭ�Ŵص�ƽ��ԭ�Ӽ����ԭ�����Ĺ�ϵ
Fig.3 Relationship between average distance and atom number of Cu nanoclusters
Ϊ�˱Ƚ�, ͼ3Ҳ�������������ʵ���ģ��(continuous elastic mediums, CEM)���۵ļ�����[16]�� CEMģ�͵Ļ�������Ϊ:�ӿ�״������ȡ��һ�����׳߶ȵ����ξ���, �൱�������˱�����, ʹ��ϵ��������, Ϊ�˽�������, ��������, ���ھ�������������һ���ĵ�����, �谭���������� �����ӵı����ܺ͵����ܴﵽƽ��ʱ, һ���ȶ����������Ͳ����ˡ� CEMģ�͵ļ��㹫ʽΪd=db[1-1/(1+(G/��)��db����-1/2��n1/3)], ʽ��d��db�ֱ�Ϊ�ŴغͿ�����ϵ�ƽ��ԭ�Ӽ��; GΪ�б�ģ��; ��Ϊ0Kʱ�ĵ�λ���������; ��Ϊ��״���ӡ� ����ͭ, db=0.2553nm[17], G=4.83GPa[18], ��=1.592mJ/m2[19], ��=1.245[15]�� �ɴ˿��Կ���, CEMģ�Ϳ����˾���ij�ԥ, ��������ͱ��о�ģ��ֵ����һ�¡� CEM�ǻ������뾧���ģ��, ���, ���ܹ�Ԥ��Ǿ��Ŵ�ƽ��ԭ�Ӽ��ı仯�� ������CEMģ�ͽ����忴��һ����������, ��û�п��Ǿ����������, ��˼��������Ŵؾ����ģ��ֵ������һ���IJ�ࡣ
���ڽϴ�ߴ��Ŵ�, һ��������ģ��: 1) ����-����ģ��, Ҳ����˵, �Ŵص��ڲ��ṹ�����û��ʲô�ֱ�, ����ֻҪ���DZ���Ӱ�����[19]; 2) ��-��ģ��, ���ŴصĽṹ���Կ������沿�ֵĿ����ڲ��ĺ������ֹ���, �ڲ��ĺ�����������ͬ, ������Ŀ���ͬ�ڿ������[20]�� ����ģ�͵�ȷ�Կ�ͨ���Զ�ż�ֲ��������о���˵���� ͼ4��ʾΪn=1055ʱ�Ķ�ż�ֲ������� ��ͼ4�ɿ���, ��ԥ�����еķ嶼������, ��ͼ1��Ҳ���������� ��˵���˳�ԥ��ľ������ԥǰ�ľ���������ͬ, ������ֻ�������ľ���������, ���������пDz�ľ����������� ����һ����������Gilbert��[21]����ZnS�ṹ��ʵ������һ�¡� ���, �����ŴصĽṹ�Ȳ��ǿ���ӱ���ṹ, Ҳ���ǿǺ˽ṹ�� ��Ҫ��ȷ�о������Ŵص�����, �������ϸ�����Ŵؽṹ�������ԡ�
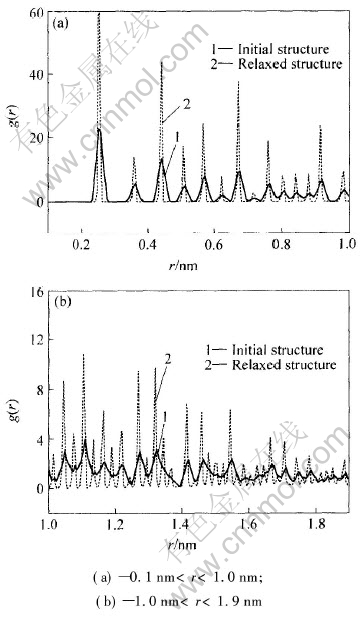
ͼ4 n=1055ʱ��ͭ�ŴصĶ�ż�ֲ�����
Fig.4 Pair correlation function of Cu clusters with n of 1055
3 ����
1) ���ųߴ�ļ�С, ͭ�����ŴصĽṹ����������Ǿ���������Ǿ���������Ǿ���ת��(n=13�Ľṹ���Կ�������Ҳ���Կ����Ǿ�)��
2) �Ŵ�ƽ��ԭ�ӽ�������ųߴ�ļ�С��ƽ���ؼ�С, ��ֻ�����ڶ̳�����, ��˲�����Ϊ�Ǿ��Ŵ��뾧̬�Ŵؽṹ�仯���оݡ�
3) ƽ��ԭ�Ӽ������Ŵؽṹ�ı仯����, �����ڳ�������, ������Ϊ�ṹ�仯��һ���оݡ�
4) ��ߴ��Ŵص��ڲ��ͱ��㶼�������ϵIJ�ͬ��
REFERENCES
[1]Heer W A D. The physics of simple metal clusters: experimental aspects and simple models [J]. Reviews of Modern Physics, 1993, 65(3): 611-676.
[2]Brack M. The physics of simple metal clusters: self-consistent jellium model and semiclassical approaches [J]. Reviews of Modern Physics, 1993, 65(3): 677-732.
[3]Valden M, Lai X, Goodman D W. Onset of catalytic activity of gold clusters on titania with the appearance of nonmetallic properties [J]. Science, 1998, 281(5383): 1647-1650.
[4]Hansen P L, Wagner J B, Helveg S, et al. Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals[J]. Science, 2002, 295(5562): 2053-2055.
[5]Binns C. Nanoclusters deposited on surfaces [J]. Surface Science Reports, 2001, 44(1-2): 1-49.
[6]Park S J, Taton T A, Mirkin C A. Array-based electrical detection of DNA with nanoparticle probes[J]. Science, 2002, 295(5559): 1503-1506.
[7]Taylor K J, Pettiette-Hall C L, Cheshnovsky O, et al. Ultraviolet photoelectron-spectra of coinage metal clusters[J]. Journal of Chemical Physics, 1992, 96(4): 3319-3329.
[8]Knickelbein M B. Electronic shell structure in the ionization potentials of copper clusters [J]. Chemical Physics Letters, 1992, 192(1): 129-134.
[9]Massobrio C, Pasquarello A, Car R. Structural and electronic-properties of small copper clusters��a first principles study[J]. Chemical Physics Letters, 1995, 238(4-6): 215-221.
[10]Kabir M, Mookerjee A, Datta R P, et al. Study of small metallic nanoparticles: an ab-initio full-potential muffin-tin orbitals based molecular dynamics study of small Cu clusters[J]. International Journal of Modern Physics B, 2003, 17(10): 2061-2075.
[11]Kabir M, Mookerjee A, Bhattacharya A K. Structure and stability of copper clusters: A tight-binding molecular dynamics study[J]. Physical Review A, 2004, 69(4): 043203.
[12]Qi W H, Wang M P. Size effect on the cohesive energy of nanoparticle[J]. Journal of Materials Science Letters, 2002, 21(22): 1743-1745.
[13]Qi W H, Wang M P, Zhou M, et al. Surface-area-difference model for thermodynamic properties of metallic nanocrystals[J]. Journal of Physics D: Applied Physics, 2005, 38(9): 1429-1436.
[14]Kittel C. Introduction to Solid State Physics[M]. New York: John & Sons Inc, 1996.
[15]Qi W H, Wang M P, Liu Q H. Shape factor of non-spherical nanoparticles[J]. Journal of Materials Science, 2005, 40(9-10): 2737-2739.
[16]Qi W H, Wang M P. Size and shape dependent lattice parameters of metallic nanoparticles[J]. Journal of Nanoparticle Research, 2005, 7(1): 51-57.
[17]Barrett C S, Massalski T B. Structure of Metals[M]. New York: Pergamon Press, 1980.
[18]Brandes E A. Smithells Metals Reference Book[M]. Boston: Butterworths, 1983.
[19]Nanda K K, Sahu S N, Behera S N. Liquid-drop model for the size-dependent melting of low-dimensional systems[J]. Physical Review A, 2002, 66(1): 013208.
[20]Palosz B, Stel��makh S, Grzanka E, et al. High pressure X-ray diffraction studies on nanocrystalline materials[J] Journal of Physics Condensed Matter, 2004, 16(5): S353-377.
[21]Gilbert B, Huang F, Zhang H, et al. Measurement of internal strain and lattice stiffening in ZnS nano-particles[J]. Science, 2004, 305(5684): 651-654.
(�༭����)
������Ŀ: ������Ȼ��ѧ����������Ŀ(50401010)
�ո�����: 2005-07-15; ������: 2005-08-20
�����: ������(1975-), ��ʿ�о���
ͨѶ����: ������, �绰: 0511-5856821; E-mail: weihong.qi@gmail.com

