


Trans. Nonferrous Met. Soc. China 22(2012) 170-174 First-principles study of TiC(110) surface
WANG Li1, FANG Li-hong2, GONG Jian-hong1
1. School of Mechanical and Electronic Engineering, Shandong University at Weihai, Weihai 264209, China;
2. Key Laboratory for Liquid-Solid Structural Evolution & Processing of Materials, Ministry of Education,
Shandong University, Ji’nan 250061, China
Received 24 February 2011; accepted 25 April 2011
Abstract: The structural and electronic properties of TiC(110) surfaces are calculated using the first-principles total-energy plane-wave pseudopotential method based on density functional theory. The calculated results of structural relaxation and surface energy for TiC(110) slab indicate that slab with 7 layers shows bulk-like characteristic interiors, and the changes of slab occur on the outmost three layers, which shows that the relaxation only influences the top three layers. Meanwhile, the strong Ti―C covalent bonding can be found in the distribution of charge density on the (100) plane. The interlayer Ti―C chemical bonds are reinforced and the outermost interlayer distance is reduced as a result of the charge depletion in the vacuum and the charge accumulations in the interlayer region between the first and second layers. The surface energy of TiC(110) is calculated to be 3.53 J/m2.
Key words: first-principles; TiC(110) surface; charge distribution; structural relaxation
1 Introduction
Like other early transition-metal carbides (TMC), TiC is characterized by a combination of properties that are traditionally assigned to two completely different types of materials: ceramics and metals [1]. TiC belongs to NaCl structure. It not only possesses many special physical properties, such as high melting point, extreme hardness, and outstanding wear resistance, but also exhibits the electric and heat conductivities, which make it highly attractive in the scientific and technological region [1]. The unique combination of these desirable physical and chemical properties led to the commercial applications of TiC as cutting tools [2], hard-coating materials [3], particular-reinforced composites [4], grain refiner [5], microelectro-mechanical systems [6], fusion- reactor walls [7], biocompatible materials [8], etc.
The experimental [9, 10] and theoretical [11-13] studies on TiC cover the fields of materials science, physics as well as chemistry. The experimental values have been listed in our previous paper of bulk and (001) surface of TiC [14]. A relatively large amount of first-principle calculation has been devoted to study the bulk properties, surface and interfacial properties of TiC.
AHUJA and ERIKSSON [13] calculated the structural and elastic properties of bulk TiC using the full potential linear muffin-tin orbital method. The phonon dispersion and phonon density of states of TiC crystal were examined by JOCHYM et al [15]. The results were compared and were in good agreement with the experimental neutron scattering data. In addition, the electronic structure and adhesion of polar TiC(111)/Ti interfaces and Al/TiC interface were also examined by LIU et al [16]. Recently, the adsorption on TiC(111) surface has been investigated with density functional calculation [1]. However, little research work was focused on TiC(110) surface or comparison of structural and electronic properties between different TiC surfaces. In this work, using the first-principles total-energy plane-wave pseudopotential method based on density functional theory (DFT), TiC(110) surfaces are investigated, focusing on their structural and electronic properties, and were compared with those of TiC(001) surface, which was already explored in Ref. [14]. In addition to its appeal from a basic science standpoint, the final motivation for the study of TiC is to investigate the grain refinement mechanism of Al-Ti-C master alloys as it is added to the Al-melts in our future work.
2 Computational details
CASTEP (Cambridge Serial Total Energy Package) software [17] was utilized in our calculations, based on density functional theory (DFT). CASTEP uses a plane wave basis set for the expansion of the single particle Kohn-Sham as implemented and ultrasoft pseudo- potentials to describe ionic cores. The minimum total energy of the structure is achieved by relaxing automatically the internal coordinates using the Broyden-Fletcher-Goldfarb-Shanno (BFGS) algorithm. The atomic configurations of Ti and C generated from the ultrasoft pseudopotential are 3s23p63d24s2 and 2s22p2, respectively. The exchange-correlation energy is described by the generalized gradient approximation of PBE (GGA-PBE). Brillouin zone sampling is performed using 8×8×1 Monkhorst-Pack k-points meshes for (110) surface calculations. The plane-wave cutoff energy in our calculations is 350 eV, which assures a total-energy convergence of 2.0×10-5 eV/atom. The diagrams of distribution of charge density and difference in charge density are obtained with the ORIGIN package.
3 Results and discussion
TiC(110) slab is constructed before the calculation by cleaving a bulk TiC after geometry optimization, as shown in Fig. 1. A vacuum region with the thickness of 20 ? is included in the supercell [18] to prevent the interaction between the slab and its periodic images. The lengths of simulated box are 4.23432 ?, 3.11618 ? and 28.7311 ?, respectively.
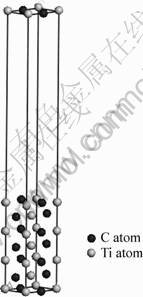
Fig. 1 Schematic diagram of TiC(110) slab with seven atomic layers
3.1 Structural relaxations and surface convergence of TiC(110) surface
Table 1 lists the results of TiC(110) surface relaxation as a function of slab thickness. It indicates that the crystal structure of TiC(110) free surface is within a very small change and surface reconstruction does not occur for (110) surface after full relaxation. It also shows that the surface relaxation is larger on the top atomic layers of the TiC slab. Moreover, it can be found that the intervals between the top and second atomic layers and between the third and forth layers are reduced except that between the third and forth atomic layers of C atoms in 7-layer slab, which are so little that they can be ignored; the intervals between the second and third layers and between the fourth and fifth layers are increased. The expansion/contraction effect does not happen on moving into deeper layers. Relaxation for (110) surface is not large, and the effect of relaxation is mainly localized within the top three atomic layers. When the slab thickness, n, is more than 7, the top three interlayers relaxation for (110) surface is well converged, which implies that the slabs with more than 7 atomic layers possess bulk-like characteristic interiors. These properties are consistent with those of (001) surface of TiC [14].
Table 1 TiC(110) surface relaxations as a function of slab thickness (change of interlayer spacing Δij as a percentage of spacing in bulk)
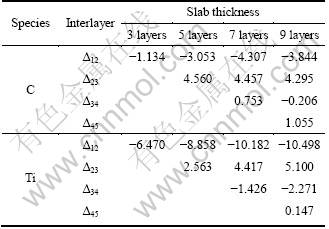
Since the properties of thin slab differ significantly from those of thick one [10], it is important to make sure that TiC slab is thick enough to show the bulk-like characteristic interiors. However, the thicker the slab is, the more complicated the computation is. Therefore, the minimum number of atomic layers should be determined to satisfy both the slabs with bulk-like characteristic interiors and lower computation cost. A preliminary analysis in Table 1 indicates that the slabs with 7 atomic layers possess the bulk-like characteristic interior. Surface energy calculation is also performed to further identify the minimum number of atomic layers. Since TiC(110) is a non-polar surface, one way to ensure the presence of a bulk-like slab is to check for the convergence of the surface energy with respect to the number of atomic layers, n [10]. Upon attaining a critical thickness, the surface energy will converge to a fixed value, indicating that the TiC slab possesses a bulk-like interior. We have calculated the surface energy of the (110) surface for the slabs of sizes ranging from 3 to 9 layers based on the method proposed by BOETTGER [19].
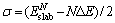 (1)
(1)
where 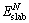 is the total energy of a N-layer slab and ΔE is the incremental energy and determined by
is the total energy of a N-layer slab and ΔE is the incremental energy and determined by 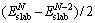 .
.
As can be seen in Table 2, the surface energy of the TiC(110) surface converges rapidly with increasing slab thickness. The calculated surface energy of TiC(110) is 3.53 J/m2 and 3.47 J/m2 for 7- and 9-layer slab, respectively, only 0.06 J/m2 difference between the two slabs, slightly higher than the difference of LIU’s calculation result with 0.04 J/m2 [10], which means that it has converged as the number of atomic layers is 7. Since the experimental value is not available in the literature, 7-layer slab of TiC(110) is chosen as further studies on the electronic structure.
Table 2 Convergence of surface energy with respect to slab thickness

3.2 Electronic structure
Table 3 lists the effective atomic charges for TiC(110) relaxed and unrelaxed surfaces using Mulliken population analysis. It is found that effective atomic charges for the atoms from the upper layers to the inner layers differ slightly after full relaxation, which implies that the charge transfer is quite small. Impossibly, small charge transfer has an effect on the distribution and types of chemical bonds between atoms in the investigated system.
Table 3 Effective atomic charges for TiC(110) surfaces
To further analyze the electronic structures of the (110) surface, the distributions of valence charge density of the relaxed surfaces and the differences in the charge densities between the relaxed and unrelaxed surfaces on the (100) plane for TiC(110) surface are shown in Fig. 2.
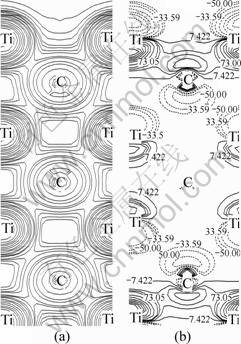
Fig. 2 Distribution of charge density (a) and difference in charge density (relative to ideal surface) (b) on (100) vertical plane for TiC(110) surface after full relaxation (Solid and dotted lines in (b) indicate positive and negative values, respectively)
It can be seen from Fig. 2(a) that strong Ti-C interaction exists between the neighboring layers, which has also been observed in our former work on bulk and (001) surface of TiC [14]. Figure 2(b) indicates that relaxations only have influence on the top three layers, while little on the interior of slabs. Charge depletion happens in the vacuum, and there are charge accumulations in the interlayer region between the first and second layers after full relaxation. It means that the interlayer Ti―C chemical bonds are reinforced and the outermost interlayer distances are largely reduced, as can be found in Table 1. However, only charge depletion exists in the region between the second and third layers, which means the chemical bonds between them are weakened, which leads to the expansion of the interlayer distance between the second and third layers, as shown in Table 1. It leads to the conclusion that charge density difference in the region between the second and third layers should be taken into account to understand the relaxation behavior of the TiC surface.
Since the (001) and (110) surfaces of TiC are both non-polar surfaces, there must be some similarities between the two surfaces: both of them show strong interaction between Ti and C atoms; the relaxation has only influence on the top three layers; and the Ti―C chemical bonds are reinforced after full relaxation. The surface energies of the two surfaces are different: 1.58 J/m2 for (001) surface and 3.53 J/m2 for (110) surface. It is impossible to investigate the grain refinement mechanism of Al-Ti-C master alloys on the Al by only the two results of TiC(001) and (110) surfaces. Further research work should be done since Al/TiC interface can provide primitive information about the substrate for the epitaxial growth of aluminum on TiC surface, which is helpful for understanding the possible nucleation mechanism of aluminum on it.
4 Conclusions
The structural and electronic properties of TiC(110) surface are explored using the first-principles total-energy plane-wave pseudopotential method based on density functional theory. The effect of relaxation is mainly localized within the top three atomic layers, and the expansion/contraction effect does not happen on moving into deeper layers. The effective atomic charges for the atoms from the upper layers to the inner layers differ slightly after full relaxation, which implies that the charge transfer is quite small. Strong Ti-C interaction exists between the neighboring layers. Charge depletion happens in the vacuum and there are charge accumulations in the interlayer region between the first and second layers after full relaxation. The interlayer distance between the second and third layers expands, which weakens Ti―C bonds between the second and third layers. The surface energy of TiC(110) calculated is 3.53 J/m2, much higher than that of (001) surface.
References
[1] RUBERTO C, LUNDQVIST B I. Nature of adsorption on TiC(111) investigated with density-functional calculation [J]. Phys Rev B, 2007, 75: 235438-1-31.
[2] TOTH L E. Transition metal carbides and nitrides [M]. New York: Academic Press, 1971.
[3] GUBANOV V A, IVANOVSKY A L, ZHUKOV V P. Electronic structure of refractory carbides and nitrides [M]. Cambridge: Cambridge University Press, 1994.
[4] PENG L M. Synthesis and mechanical properties of niobium aluminide-based composites [J]. Materials Science and Engineering A, 2008, 480: 232-236.
[5] WANG Z Q, LIU X F, LIU Y H, ZHANG J Y, YU L N, BIAN X F. Structural heredity of TiC and its influences on refinement behaviors of AlTiC master alloy [J]. Transactions of Nonferrous Metals Society of China, 2003, 13: 790-793.
[6] SONG M S, HUANG B, ZHANG M X, LI G J. Study of formation behavior of TiC ceramic obtained by self-propagating high- temperature synthesis from Al-Ti-C elemental powders [J]. Int Journal of Refractory Metals & Hard Materials, 2009, 27: 584-589.
[7] HOJOU K, OTSU H, FURUNO S, SASAJIMA N, IZUI K. In situ observation of damage evolution in TiC during hydrogen and deuterium ion irradiation at low temperatures [J]. Journal of Nuclear Materials, 1996, 239: 279-283.
[8] JONES M I, MCCOLL I R, GRANT D M, PARKER K G, PARKER T L. Protein adsorption and platelet attachment and activation, on TiN, TiC, and DLC coatings on titanium for cardiovascular applications [J]. Journal of Biomedical Materials Research, 2000, 54: 413-421.
[9] DUNAND A, FLACK H D, YVON K. Bonding study of TiC and TiN (I). High-precision X-ray diffraction determination of the valence-electron density distribution, Debye-Waller temperature factors, and atomic static displacements in TiC0.94 and TiN0.99 [J]. Phys Rev B, 1985, 31: 2299-2315.
[10] LIU L M, WANG S Q, YE H Q. Adhesion and bonding of the Al/TiC interface [J]. Surface Science, 2004, 550: 46-56.
[11] DUDIY S V, LUNDQVIST B I. First-principles density-functional study of metal-carbonitride interface adhesion: Co/TiC(001) and Co/TiN(001) [J]. Phys Rev B, 2001, 64: 045403-1-14.
[12] ARYA A, CARTER E A. Structure, bonding, and adhesion at the TiC(100)/Fe(110) interface from first principles [J]. The Journal of Chemical Physics, 2003, 118: 8982-8996.
[13] AHUJA R, ERIKSSON O. Structural, elastic, and high-pressure properties of cubic TiC, TiN, and TiO [J]. Phys Rev B, 1996, 53: 3072-3079.
[14] FANG L H, WANG L, GONG J H, DAI H S, MIAO D Z. First-principles study of bulk and (001) surface of TiC [J]. Transactions of Nonferrous Metals Society of China, 2010, 20: 857-862.
[15] JOCHYM P T, PARLINSKI K, STERNIK M. TiC lattice dynamics from ab initio calculations [J]. Eur Phys J B, 1999, 10: 9-13.
[16] LIU L M, WANG S Q, YE H Q. First-principles study of the polar TiC/Ti interface [J]. J Mater Sci Technol, 2003, 19: 540-544.
[17] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Cond Matt, 2002, 14: 2717-2744.
[18] HAN Y F, DAI Y B, SHU D, WANG J, SUN B D. First-principles study of TiB2(0001) surfaces [J]. J Phys: Condens Matter, 2006, 18: 4197-4205.
[19] BOETTGER J C. Nonconvergence of surface energies obtained from thin-film calculations [J]. Phys Rev B, 1994, 49: 16798-16800.
TiC(110)表面第一性原理研究
王 丽1,房立红2,宫建红1
1. 山东大学 威海校区机电工程学院,威海 264209;
2. 山东大学 材料液固结构演变与加工教育部重点实验室,济南 250061
摘 要:利用基于密度泛函理论的第一性原理平面波赝势法计算TiC(110)表面的结构和电子特性。对于该表面结构弛豫和表面能的计算结果表明,7层原子构型能够显示TiC的内部体相特征,弛豫后结构的变化仅发生在顶部3层,证明弛豫只影响构型的顶部3层。同时,从构型的(100)平面上的电荷密度分布中可以看到强烈的Ti―C共价键。弛豫后,由于电荷在真空层中的消耗和第一、第二层原子层之间的积累,第一、第二层原子间距减小, Ti―C化学键相应地增强。计算得到的TiC(110)表面的表面能为3.53 J/m2。
关键词:第一性原理;TiC(110)表面;电荷分布;结构弛豫
(Edited by YUAN Sai-qian)
Foundation item: Project (200902554) supported by National Post-doctor Foundation, China; Project (200802015) supported by the Post-Doctor Foundation of Shandong Province, China
Corresponding author: WANG Li; Tel: +86-631-5682092; E-mail: wanglihxf@sdu.edu.cn
DOI: 10.1016/S1003-6326(11)61157-6

