���±�ţ�1004-0609(2013)08-2147-09
���ڵ�һ��ԭ��L12-Al3Sc��ȱ�ݽṹ���ɼ���Ϊ�ļ���
��˳ƽ1����Сƽ1��������1�������1�����Ͳ�2��������2��������1���� ��3������3
(1. ��������ѧԺ ���Ϲ���ѧԺ������ 213001��
2. ����ѧԺ ��е����ӹ���ϵ������ 247000��
3. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410083)
ժ Ҫ�����õ�һ��ԭ��ƽ�沨���Ʒ�����������仯����L12-Al3Sc�Ļ������ԣ���ͨ�������ȱ���γ����Ʋ�L12-Al3Sc��ȱ�ݵ���Ҫ������ʽ����ϵ���ܶȺ�̬�ܶȵķ�����ʾL12-Al3Sc�ijɼ���Ϊ�����������L12-Al3Sc�ľ�����Ϊ4.107 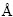 ����ģ��Ϊ86.5 GPa���γ���Ϊ-43.83 kJ/mol��L12-Al3Sc�ĵ�ȱ����ҪΪAl�Ǿ����ϵ�Al��λ��Sc��λȱ�ݡ�L12-Al3Sc��Sc��λ��Al��λȱ�ݵ��γ��ܽ�Ϊ�ӽ���������Al�Ͻ���Sc��λ��Al��λȱ�����ڹ�ͬ���ڣ�Sc��λȱ�ݵ��γ���С��Al��λ�ģ�������Sc�Ͻ�ĵ�ȱ��ΪSc��λȱ�ݡ�L12-Al3Sc�ijɼ�����ܶȳʷĴ�״�����ֳ�Sc d-Al p�Ĺ���ӻ�ЧӦ�����ӻ������ҪΪSc dz2-Al pz����ӻ���
����ģ��Ϊ86.5 GPa���γ���Ϊ-43.83 kJ/mol��L12-Al3Sc�ĵ�ȱ����ҪΪAl�Ǿ����ϵ�Al��λ��Sc��λȱ�ݡ�L12-Al3Sc��Sc��λ��Al��λȱ�ݵ��γ��ܽ�Ϊ�ӽ���������Al�Ͻ���Sc��λ��Al��λȱ�����ڹ�ͬ���ڣ�Sc��λȱ�ݵ��γ���С��Al��λ�ģ�������Sc�Ͻ�ĵ�ȱ��ΪSc��λȱ�ݡ�L12-Al3Sc�ijɼ�����ܶȳʷĴ�״�����ֳ�Sc d-Al p�Ĺ���ӻ�ЧӦ�����ӻ������ҪΪSc dz2-Al pz����ӻ���
�ؼ��ʣ�L12-Al3Sc����ȱ�ݽṹ����һ��ԭ�����㣻�ɼ���Ϊ���ӻ�
��ͼ����ţ�TG146.2 �� �� ���ױ�־�룺A
Calculation of point defect structures and bonding behavior of L12-Al3Sc intermetallic based on first-principles
SUN Shun-ping1, LI Xiao-ping1, LEI Wei-ning1, WANG Hong-jin1, WANG Xian-cai2, JIANG Hai-feng2, LI Ren-xing1, JIANG Yong3, YI Dan-qing3
(1. School of Materials Engineering, Jiangsu University of Technology, Changzhou 213001, China;
2. Department of Mechanics and Electronic Engineering, Chizhou University, Chizhou 247000, China;
3. School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: The first-principles pseudopotential plane-wave calculation was used to study the energies and the electronic properties of point defects for L12-Al3Sc intermetallic. According to the calculation and comparison of the formation energy of point defect structures, the geometrical configuration of point defects in L12-Al3Sc intermetallic was analyzed. Combining with densities of states and charge densities, the effect of bonding behavior on electronic structure was investigated emphatically. The results show that the lattice constant of L12-Al3Sc is 4.107 ��bulk modulus is 86.5 GPa, and formation enthalpy is -43.83 kJ/mol. These calculation results also suggest that the point defects in the Al sublattices, i.e., Al vacancy and Sc anti-site, are more favorable than those in the Sc sublattices for the stoichiometric compound Al3Sc in the L12 structure, but Sc vacancy and Al anti-site defect coexist in rich-Al alloy, and mainly Sc anti-site defect can be found in rich-Sc alloy. The Al vacancy-induced charge density shows the spindle-like bonding characteristic, and Sc dz2-Al pz orbital hybridization is depicted.
Key words: L12-Al3Sc; point defect structures; first-principles calculation; bonding behavior; hybridization
���ֺϽ������ܶȵͣ��Ҿ��нϸߵ�����ǿ�Ⱥ�����ĸ��¿�������ܣ��Ϻõ����Ƶڶ���ֻ�����������һ�ּ���ǰ����ʱЧǿ���������Ͻ��ں��պ�������������Ҫ��Ӧ��[1-4]��ϡ��Ԫ����Ҳ����Ϊ�����Ͻ���ǿ��Ч����õĺϽ�Ԫ�ء�Sc�����Ͻ��п��γ�Al3Sc�࣬L12�ṹ���������徧�����С(1.34%)���߶���ɢ�����ȷֲ����������С���Al3Sc��ߴ�ϸС��Ϊ20~30 nm�����������γɹ�����棬�������Ni�����ºϽ���Ni/�á�-Ni3Al�������ƣ�������ǿ��Ч���ܺá�����Al3Sc�۵��(1 320 ��)�����ȶ��Ժã���ǿ�������ȼӹ��������ٽᾧ������ٽᾧ�¶ȣ����ƺϽ�ĸ������ܡ�
Al3Sc����Ϊ���ֺϽ��е���Ҫ�����࣬�й�����ò���ṹ�����������Լ��������е��о��Ѿ���չ�˺ܶࡣ����������Ԫ����ǿ����Ϊ���Ͻ��о����ȵ�����֮һ[5-10]�����ֺϽ��и���������ķ����ٴμ��������Ƕ�Al3Sc��Ĺ�ע���о����ָ�����������γ��벻ͬ������֮�������������Լ�������ĵ�ȱ����Ϊ�ܲ��ɷ�[11-12]��������L12-Al3Sc��ȱ�ݽṹ��������չ�о�������Ҫ����ʵ����[11-15]��FAZELI��[14]ָ����ʣ�Ŀ�λ��Al-Mg-Sc�Ͻ����������������Ҫ���ã������Kampmann�CWagner (KWN)ģ�Ͳ��Ϳ�λ��Al-Mg-Sc�Ͻ��е����û�����WOODWARD��[15]��ͨ����һԭ������L12-Al3Sc�ĵ�ȱ��Ũ�ȡ�
��������ͨ����һ��ԭ������L12-Al3Sc�����仯����Ļ������Լ���ȱ�ݽṹ����ͨ����ȱ���γ��ܵļ����Ʋ�L12-Al3Sc��ȱ�ݵ���Ҫ������ʽ���ڴ˻����ϣ�����L12-Al3Sc�ĵ���ܶȡ��ɼ�����ܶȼ�̬�ܶȣ���ͨ���Ե�ȱ�ݽṹ���о�L12-Al3Sc�ijɼ���Ϊ��
1 ���㷽��
���еĵ�һ��ԭ�����������VASP (Vienna Ab-inito Simulation Package)���ܼ������������[16]���С�VASP ����ļ����ǻ����ܶȷ������۵ĵ�һԭ��ƽ�沨���Ʒ��������еġ��ڽ� ����ʱ������Al��Sc���������Ʋ��þֲ��ܶȽ���(LDA-CA[17])�����ݶȽ���(GGA-PW91[18]��GGA-PBE[19])��ѡ��ͶӰ��ƽ�沨����(PAW)[20]����ȷ��������-����֮�������ã�����Al��ScԪ�طֱ�Al 2s2p��Sc 3s3p4s3d��Ϊ�۵��ӣ������ڲ�ԭ����Ϊо���ӡ�����ģ�͵ļ�Լ����Ԩ����K���������MONKHORST-PACK[21]����(18��18��18)�����֡�����BLOCHL��[22]������Linear-Tetrahedron��������ȷ�ؼ���ϵͳ���ܡ����������Ա߽��������侧���е���Kohn-Sham��������ƽ�沨����(PW)չ����ƽ�沨��Ŀ�ɶ��ܽضϵ�������������Al��Sc��L12-Al3Sc��ԭ�������ضϵ�ѡȡΪ320 eV��
����ʱ������Al��Sc���������Ʋ��þֲ��ܶȽ���(LDA-CA[17])�����ݶȽ���(GGA-PW91[18]��GGA-PBE[19])��ѡ��ͶӰ��ƽ�沨����(PAW)[20]����ȷ��������-����֮�������ã�����Al��ScԪ�طֱ�Al 2s2p��Sc 3s3p4s3d��Ϊ�۵��ӣ������ڲ�ԭ����Ϊо���ӡ�����ģ�͵ļ�Լ����Ԩ����K���������MONKHORST-PACK[21]����(18��18��18)�����֡�����BLOCHL��[22]������Linear-Tetrahedron��������ȷ�ؼ���ϵͳ���ܡ����������Ա߽��������侧���е���Kohn-Sham��������ƽ�沨����(PW)չ����ƽ�沨��Ŀ�ɶ��ܽضϵ�������������Al��Sc��L12-Al3Sc��ԭ�������ضϵ�ѡȡΪ320 eV��
�����õ�һ��ԭ�����������ȱ�ݽṹʱ����Ҫ������ԭ����һ����Ϊ����ԭ��ģ�ͳߴ�Ĵ�С�Ե�ȱ�ݼ�������Ӱ����Խ�С�����ڼ�������е��ռ�����Ļ���(K���ȡֵ)�Լ���Ľ����Ӱ�첻�ݺ���[23]�������ԭ��ģ�͵Ĵ�С�͵��ռ�����Ļ���Ӧ����ȡֵ�������Լ��㣬����ȷ����L12-Al3Sc��ȱ�ݽṹ�����ù���2��3��3 ��ԭ���ķ��������о����䵹�ռ�����Ļ���Ϊ9��6��6������Broyden-Flecher-Goldfarb-Shanno(BFGS)����[24]�Ը��ֳ�ԭ��ģ�ͽ����˼����Ż�����������ǵľ������ȶ��ṹ��������Ǣ����(SCF)ʱ��Ӧ����PULAY[25]����㷨�����㣬��ϵ������������ȡֵΪ5.0��10-6 eV/atom��ÿ��ԭ��������0.1 eV/nm������ƫ��С��5��10-5 nm��Ӧ��ƫ�����0.02 GPa��L12-Al3Sc�����ֵ�ȱ�ݵĽṹʾ��ͼ��ͼ1��ʾ��
2 ������������
2.1 Al��Sc��L12-Al3Sc�Ļ�������
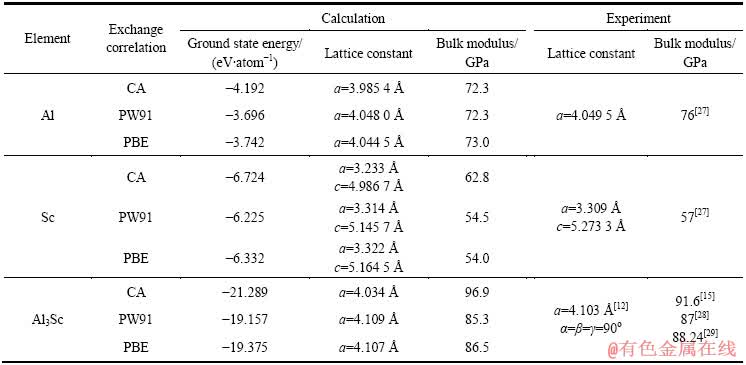
���ȼ�������仯����L12-Al3Sc������ԪAl��Sc��Ԫ�صĻ������ʣ�����������1����Ԫ�صĻ�̬��������ģ���;������ȿ�ͨ�����E��V���߽�Murnaghan[26]״̬���̵õ���״̬��������
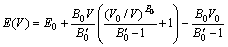 (1)
(1)
ʽ�У�V��E(V)�ֱ�Ϊ���������Ͷ�Ӧ��������V0��E0��B0�ֱ�Ϊ��̬�������̬�����Լ����ϵ���ģ���� Ϊ������������ֵһ����3.5~5.5֮�䡣
Ϊ������������ֵһ����3.5~5.5֮�䡣
�ӱ�1�п��Կ��������Al��ScԪ�صľ�������ʵ��ֵ���ϵĺܺã�Ԥ�⾫���������⡣
�ڴ�Ԫ�صļ���֮���ֶ�L12-Al3Sc���м��㣬�������Ҳ���ڱ�1�С��ӱ�1�п��Կ��������ø������Ƽ���õ���L12-Al3Sc�ľ�������ʵ��ֵ����������1%���ڣ���ģ�����������10%���ڡ����У�ͨ��PBE���Ƽ���õ��ľ�����a=4.107 ����ģ��B0=86.5 GPa����������ʵ��ֵ���ϵúܺá�
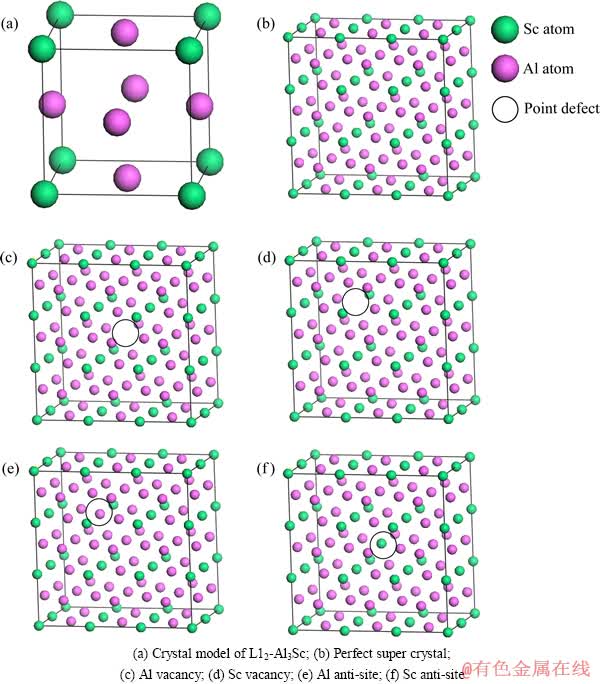
ͼ1 L12-Al3Sc��ȱ�ݽṹ����ģ��
Fig. 1 Calculation models of point defect structures of L12-Al3Sc crystal
��1 Al��Sc��L12-Al3Sc�ĵ�һԭ������ֵ��ʵ��ֵ
Table 1 First-principles calculations and experimental data of Al, Sc and L12-Al3Sc
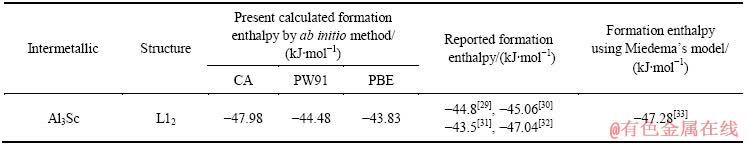
��2 L12-Al3Sc�γ��ʵĵ�һԭ������ֵ�ͱ���ֵ
Table 2 Calculations and reported data of formation enthalpies of L12-Al3Sc
��������γ�����ָԭ���ɵ���״̬�γɻ�����ʱ�ͷŵ���������������������������γ����������ǿ�ȼ���ѧ�ȶ��ԡ�һ����ԣ��γ���Ϊ��ֵ���Ҿ���ֵԽ��������û�����Ľ������Խǿ���ȶ���Խ�ߡ�L12-Al3Sc�γ��ʵļ��㹫ʽ���£�
 (2)
(2)
ʽ�У�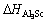 ΪL12-Al3Sc���γ��ʣ�
ΪL12-Al3Sc���γ��ʣ� ΪL12-Al3Sc����������������EAl��ESc�ֱ�ΪAl��Sc�ȶ����ʾ����е���ԭ�ӵ�������
ΪL12-Al3Sc����������������EAl��ESc�ֱ�ΪAl��Sc�ȶ����ʾ����е���ԭ�ӵ�������
L12-Al3Sc�γ��ʵļ��������ڱ�2�С��ӱ�2�п������ø������Ƽ���õ��ĵļ����������˵�ʵ������۱���ֵ������10%���¡�ͨ��PBE���Ƽ���õ����γ���=-43.83 kJ/mol���������������⡣�ɱ�1��2�ɼ���PBE���Ƽ���õ���L12-Al3Sc�Ļ������Բ����뱨��ֵ�����ϵúܺã�����ں����ĵ�ȱ�ݽṹ���ɼ���Ϊ�о��У�ֻ����GGA-PBE���ƽ��м��㡣
2.2 L12-Al3Sc�ĵ�ȱ���γ���
L12-Al3Sc��Al��ScԪ�صĿ�λ�γ���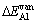 ��
��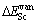 �����㹫ʽ����
�����㹫ʽ����
 (3a)
(3a)
 (3b)
(3b)
ʽ�У� ��
�� �ֱ�ΪL12-Al3Sc��Al��ScԪ�صĿ�λ�γ��ܣ�
�ֱ�ΪL12-Al3Sc��Al��ScԪ�صĿ�λ�γ��ܣ�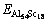 ΪAl 54Sc18��ԭ������������
ΪAl 54Sc18��ԭ������������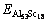 ��
��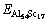 �ֱ�ΪAl 53Sc18��Al 54Sc17��ԭ������������
�ֱ�ΪAl 53Sc18��Al 54Sc17��ԭ������������
L12-Al3Sc��Al��ScԪ�صķ�λȱ���γ��� ��
��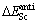 �����㹫ʽ����
�����㹫ʽ����
 (4a)
(4a)
 (4b)
(4b)
ʽ�У� ���ֱ�ΪL12-Al3Sc��Al��ScԪ�صķ�λȱ���γ��ܣ� ��
�� �ֱ�ΪAl55Sc17��Al53Sc19��ԭ������������
�ֱ�ΪAl55Sc17��Al53Sc19��ԭ������������
ͨ��ʽ(3)��(4)�ɼ���L12-Al3Sc��λ�ͷ�λȱ�ݵ��γ��ܣ�����������3��
��3 L12-Al3Sc��λ�ͷ�λȱ���γ��ܵĵ�һԭ������ֵ
Table 3 Formation energy of vacancies and anti-sites of L12-Al3Sc system calculated by ab initio method

�ӱ�3�п��Կ�����L12-Al3Sc��Sc��λ�γ��ܸ���Al��λ������ζ����L12-Al3Sc��Al��λ�����γɣ���Al��λȱ�ݵ��γ��ܶ�Ҫ����Sc��λ�ģ��������L12-Al3Sc��Sc��λȱ�ݸ����γɡ������L12-Al3Sc�ĵ�ȱ����Ҫ��Al�Ǿ���ȱ�ݡ�
ƫ��L12-Al3Sc���뻯ѧ�����ȵĸ�Al�Ͻ���ܳ��ֵĽṹȱ��ΪAl��λȱ�ݺ�Sc��λ�� L12-Al3Sc��Sc��λ�γ�����Al��λȱ���γ��ܽ�Ϊ�ӽ���Sc��λ�γ�����ֵ��С��������ڸ�Al�Ͻ���Sc��λ��Al��λȱ�����ڹ�ͬ���ڣ���������Sc��λΪ����������ƫ�����뻯ѧ�����ȵĸ�Sc�Ͻ���ܳ��ֵĵ�ȱ��ΪSc��λȱ�ݺ�Al��λ��Sc��λȱ�ݵ��γ���С��Al��λ��������Sc�Ͻ�ĵ�ȱ��ΪSc��λȱ�ݡ���Ȼ��������Ǵ�ȱ���γ��ܳ�����һ�������жϡ�
ȱ�ݽṹ��̬�ܶ��������ķ�ӳȱ������������·ֲ������̬�ֲ��������������ֲ����ȱ�ݵ��ȶ�������Ҫ����ϵ��L12-Al3Sc��ȱ�ݽṹ��̬�ܶȼ�������ͼ2��ʾ��
��ͼ2(a)���Կ�����Sc��λ��Al��λ�ɼ�������λ�ý�Ϊ�ӽ����������Al�Ͻ���Sc��λ��Al��λȱ�����ڹ�ͬ���ڣ���������ע��ڵ��ڷ����ܵĽϸ���������Sc��λ�ijɼ��������Al��λȱ���������������ƫ�ƣ���Ҳ��ʾ��Sc��λ�ںϽ��и�Ϊ�ȶ������ּ��ʸ��ߡ���ͼ2(b)���Կ�����Sc��λȱ�ݵijɼ��������Al��λ������������ƶ����������Sc�Ͻ���Sc��λȱ�ݸ�Ϊ�ȶ����ɴ˿ɼ���̬�ܶȵļ�����������ͨ���γ��ܼ����Ʋ�Ľ������һ�¡�
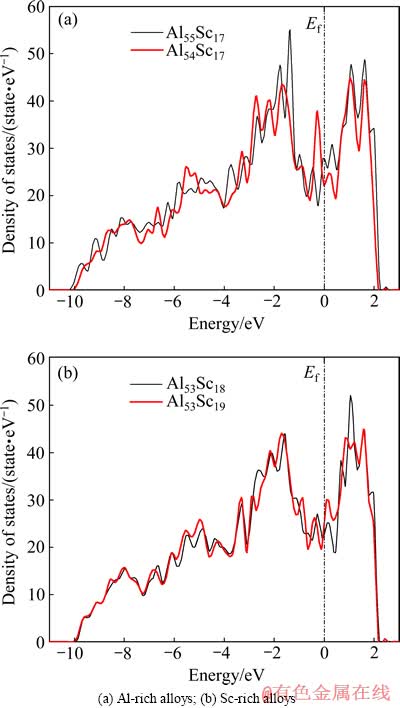
ͼ2 L12-Al3Sc��ȱ�ݽṹ��̬�ܶ�
Fig. 2 Density of states of L12-Al3Sc with point defects
2.3 L12-Al3Sc�ĵ�ȱ�ݽṹ
��ֵ���ܶ��������ط�ӳ��ȱ������������·ֲ����ԭ��֮��ĵ���ƶ��Լ��ɼ�������������ʡ���ֵ���ܶȶ���Ϊ�ɼ�ǰ����ת�Ƶĵ���ܶȲ��ʽ���£�
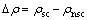 (5)
(5)
ʽ�У� Ϊ��ֵ���ܶȣ�
Ϊ��ֵ���ܶȣ� Ϊ��Ǣ�ĵ���ܶȣ�
Ϊ��Ǣ�ĵ���ܶȣ�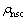 Ϊ����Ǣ�ĵ���ܶȡ�
Ϊ����Ǣ�ĵ���ܶȡ�
�����㣬L12-Al3Sc��������ĵ���ܶȺͲ�ֵ���ܶ���ͼ3��ʾ��
��ͼ3(a)�ĵ���ܶ�ͼ�п��Կ�����������L12-Al3Sc�ĵ���û�����Եľ����ԣ����־��������������ἰ�ij������������еġ����Ӻ����ص㡣���� Sc�������п�����3s��3p��4s��3d��11�����ӣ���Al�������н�������3s��3p��3�����ӣ������Scԭ�Ӻ�Alԭ����ͼ3���������֣�Scԭ���������ܶȽϸߣ�Alԭ���������ܶȽϵ͡���ͼ3(b)�IJ�ֵ���ܶ�ͼ�п��Կ�����Scԭ�ӵĵ��ӵ���Alԭ�ӷ�����ת�ƣ���Alԭ�Ӻ�Scԭ�ӵĵ���֮����ֳ���ǿ���ӻ�ЧӦ�����ֳ����ۼ����ص㣬�����L12-Al3Sc��ԭ�ӵĽ�Ϸ�ʽ�ǽ������빲�ۼ���ͬ���ڵĻ�ϼ�����һ�������Ͻ����仯�����һ����ɡ�
ͼ4��ʾΪL12-Al3Sc���ֵ�ȱ�ݽṹ�IJ�ֵ���ܶȡ���ͼ4�пɿ�������λ�ij��������˵�ɵ����·ֲ�����λ���������Եĵ���ܶȷֲ������ǽ�������λ��Χԭ�����λ�����ƶ�������ԭ�ӳ�ԥ��ͬ���أ���λȱ�ݵĴ���Ҳʹ�õ�ɷ���ת�Ʋ����յ���ԭ�ӳ�ԥ����Щ��ȱ�ݵĴ��ڽ����¾������IJ��������L12-Al3Sc�Ļ������Բ���Ӱ�졣
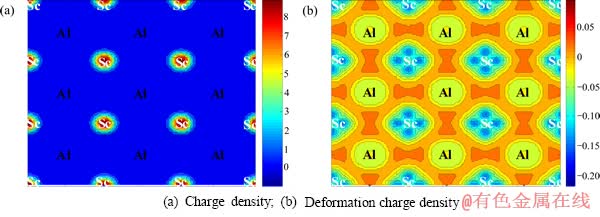
ͼ3 L12-Al3Sc��������ĵ���ܶ�ͼ����ֵ���ܶ�ͼ((100)����)
Fig. 3 Charge density and deformation charge density of L12-Al3Sc perfect crystal (on (100) plane)
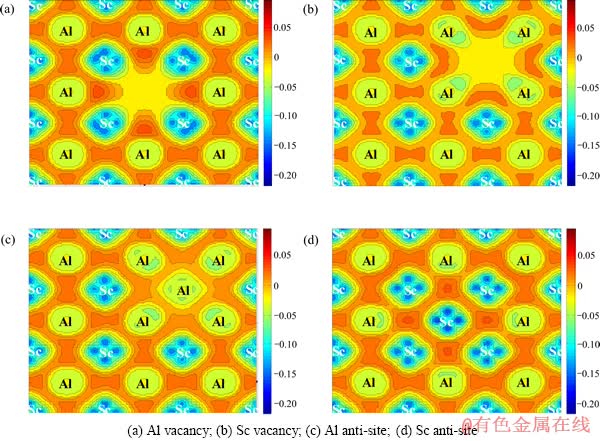
ͼ4 L12-Al3Sc��ȱ�ݽṹ�IJ�ֵ���ܶ�ͼ((100)����)
Fig. 4 Deformation charge density of L12-Al3Sc with point defects (on (100) plane)
2.4 L12-Al3Sc�ijɼ���Ϊ
Ϊ�˸��õ��о���ȱ�ݶ���L12-Al3Sc�Ͻ���Ϊ��Ӱ�죬���Խ�һ���о���ȱ���������ܶȵ����·ֲ����������ܶȵ����·ֲ�����ͨ��ȱ�ݾ��������������ֵ���ܶȵIJ�ֵ( )����ʾ����ʽ����
)����ʾ����ʽ����
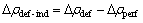 (6)
(6)
ʽ�У� Ϊȱ�ݾ���IJ�ֵ���ܶȣ���
Ϊȱ�ݾ���IJ�ֵ���ܶȣ���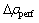 Ϊ��������IJ�ֵ���ܶȡ�
Ϊ��������IJ�ֵ���ܶȡ�
L12-Al3Sc�Ͻ��У�����Al��λ��ʽ(6)�ɼ�Ϊ
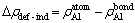 (7)
(7)
��ʽ(7)�п��Կ������ܹ���ӳAlԭ������Scԭ���γ�Al3Sc�����仯����ʱ�ijɼ�����������ɶ���Ϊ��Al��λ����ijɼ�����ܶ�[34]�����ڷ���L12-Al3Sc�ĺϽ���Ϊ������Ҫ�����壬�����L12-Al3Sc�ijɼ�����ܶ�ͼ(ͼ5)��
��ͼ5�п��Կ���Alԭ���ڳɼ������е����ܶȵı仯�����Alԭ����Scԭ�ӵĹ���ӻ�ЧӦ����������ʾ���������ַĴ�״�����ֳ�Sc d-Al p�ӻ�ЧӦ�����ݳɼ�����ܶȵ���̬���ɽ�һ���ж����ӻ������Ҫ��Sc dz2-Al pz����ӻ���d-p������ӻ�ЧӦ�����ŵ���ܶȵ����·ֲ���ԭ��֮��ļ�������ǿ����Ҳ�ǽ����仯�������������Ԫ����ǿ�Ⱥ��۵���ߵ�ԭ��
�ֲ�̬�ܶ���ֱ�۵ظ����������ϳɼ�ʱ�Ĺ�����ԣ��������ͨ����L12-Al3Sc�ֲ�̬�ܶȽ����о���ʾ��ɼ�������L12-Al3Sc��̬�ܶȼ�������ͼ6��ʾ��
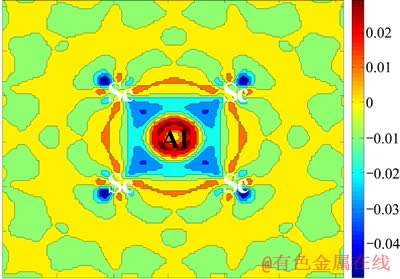
ͼ5 L12-Al3Sc Al��λ����ijɼ�����ܶ�((100)����)
Fig. 5 Al vacancy-induced charge density of L12-Al3Sc (on (100) plane)
��ͼ6�п��Կ�����L12-Al3Sc��Alԭ�Ӻ�Scԭ�ӷ������ϵ�̬�ܶȾ���Ϊ0��̬�ܶ�������������壬���Դ�������϶���ⶼ���ֳ������仯�����������Ĺ��ۼ���������һ�����ǰ���IJ�ֵ���ܶȵĽ��(ͼ3(b))һ�¡���ͼ6(a)��(b)�Ͽ��Կ�����Al��p������Sc��d���ӵ�̬�ܶȳ��ֹ���˵��Al��p������Sc��d���Ӳ�����ǿ������ã�����Ҫ��Sc d-Al p����ӻ��Ĺ��ס�ͨ����һ���Ա�ͼ6(c)��(d)�ɵã�Sc d-Al p����ӻ��ľ����������Ƚϵ����ս����ʾ��ͼ6(e)�С���ͼ6(e)�п��Կ�����L12-Al3Sc Sc d-Al p�ӻ���Ҫ��Sc dz2-Al pz����ӻ��Ĺ��ף���һ�����ɼ�����ܶȵĽ��(��ͼ5)�Ǻϡ�
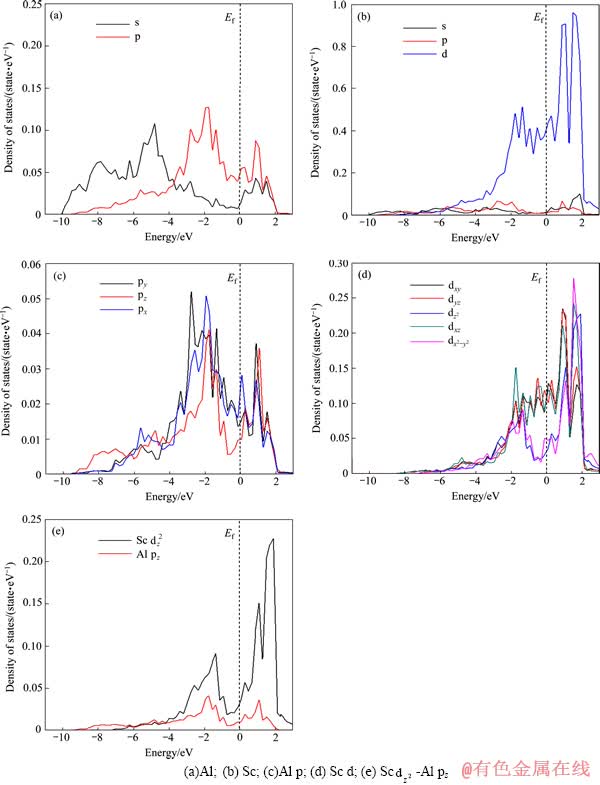
ͼ6 L12-Al3Sc��̬�ܶ�
Fig. 6 Density of states of L12-Al3Sc
3 ����
1) �����ܶȷ���ƽ�沨���Ʒ�����ѡ��PBE���Ƽ���õ�L12-Al3Sc�ľ�����a=4.107 ����ģ��B0=86.5 GPa���γ��� =-43.83 kJ/mol�������Բ������뱨��ֵ���ϵúܺá�
=-43.83 kJ/mol�������Բ������뱨��ֵ���ϵúܺá�
2) L12-Al3Sc�ĵ�ȱ����Ҫ��Al�Ǿ����ϵ�Al��λ��Sc��λȱ�ݡ� L12-Al3Sc��Sc��λ�γ�����Al��λȱ���γ��ܽ�Ϊ�ӽ���������Al�Ͻ�Sc��λ��Al��λȱ�����ڹ�ͬ���ڣ�Sc��λȱ�ݵ��γ��ܵ���Al��λ�ģ�������Sc�Ͻ�ĵ�ȱ��ΪSc��λȱ�ݡ�
3) L12-Al3Sc�ijɼ�����ܶȳʷĴ�״�����ֳ�Sc d-Al p�Ĺ���ӻ�ЧӦ�����ӻ������ҪΪSc dz2-Al pz����ӻ���
REFERENCES
[1] KARNESKY R A, DUNAND D C, SEIDMAN D N. Evolution of nanoscale precipitates in Al microalloyed with Sc and Er[J]. Acta Materialia, 2009, 57(14): 4022-4031.
[2] KRUG M E, DUNAND D C, SEIDMAN D N. Effects of Li additions on precipitation-strengthened Al-Sc and Al-Sc-Yb alloys[J]. Acta Materialia, 2011, 59(4): 1700-1715.
[3] �� ��, ������, �� ӭ, �� ��, ��־Ұ, ��־��. ��Sc��Zr��Al-Mg-Mn�Ͻ���֯����ѧ���ܵ�Ӱ��[J]. �й���ɫ����ѧ��, 2012, 22(6): 1555-1563.
CHEN Qin, PAN Qing-lin, WANG Ying, PENG Hong, ZAHNG Zhi-ye, YIN Zhi-min. Effects of minor scandium and zirconium on microstructure and mechanical properties of Al-Mg-Mn alloys[J]. The Chinese Journal of Nonferrous Metals, 2012, 22(6): 1555-1563.
[4] ����Ԫ, �ij���, ������, ������. Al-Zn-Mg-Cu-Zr-Sc�Ͻ���̬Al3(Sc,Zr)����ò���о�[J]. ϡ�н��������빤��, 2011, 40(2): 265-268.
DAI Xiao-yuan, XIA Chang-qing, LONG Chun-guang, KOU Li-li. Morphology of primary Al3(Sc,Zr) of as-cast Al-Zn-Mg-Cu-Zr-Sc alloys[J]. Rare Metal Materials and Engineering, 2011, 40(2): 265-268.
[5] COSTA S, PUGA H, BARBOSA J, PINTO A M P. The effect of Sc additions on the microstructure and age hardening behaviour of as cast Al-Sc alloys[J]. Materials and Design, 2012, 42: 347-352.
[6] DESCHAMPS A, LAE L, GUYOT P. In situ small-angle scattering study of the precipitation kinetics in an Al-Zr-Sc alloy[J]. Acta Materialia, 2007, 55(8): 2775-2783.
[7] LEFEBVRE W, DANOIX F, HALLEM H, FORBORD B, BOSTEL A, MARTHINSEN K. Precipitation kinetic of Al3(Sc,Zr) dispersoids in aluminium[J]. Journal of Alloys and Compounds, 2009, 470(1/2): 107-110.
[8] LOHAR A K, MONDAL B, RAFAJA D, KLEMM V, PANIGRAHI S C. Microstructural investigations on as-cast and annealed Al-Sc and Al-Sc-Zr alloys[J]. Materials Characterization, 2009, 60(11): 1387-1394.
[9]  M, PONGRATZ P, DEGISCHER H P. Coherency loss of Al3(Sc,Zr) precipitates by deformation of an Al-Zn-Mg alloy[J]. Acta Materialia, 2012, 60(10): 4247-4254.
M, PONGRATZ P, DEGISCHER H P. Coherency loss of Al3(Sc,Zr) precipitates by deformation of an Al-Zn-Mg alloy[J]. Acta Materialia, 2012, 60(10): 4247-4254.
[10] JIA Zhi-hong,  J, SOLBERG J K, LIU Qing. Formation of precipitates and recrystallization resistance in Al-Sc-Zr alloys[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(8): 1866-1871.
J, SOLBERG J K, LIU Qing. Formation of precipitates and recrystallization resistance in Al-Sc-Zr alloys[J]. Transactions of Nonferrous Metals Society of China, 2012, 22(8): 1866-1871.
[11] BOOTH-MORRISON C, MAO Z, DIAZ M, DUNAND D C, WOLVERTON C, SEIDMAN D N. Role of silicon in accelerating the nucleation of Al3(Sc,Zr) precipitates in dilute Al-Sc-Zr alloys[J]. Acta Materialia, 2012, 60(12): 4740-4752.
[12] MAO Z, CHEN W, SEIDMAN D N, WOLVERTON C. First-principles study of the nucleation and stability of ordered precipitates in ternary Al-Sc-Li alloys[J]. Acta Materialia, 2011, 59(8): 3012-3023.
[13] WANG Ren-nian, TANG Bi-yu, PENG Li-ming, DING Wen-jing. Ab initio study of the effect of Zr content on elastic and electronic properties of L12-Al3(Sc1-xZrx) alloys[J]. Computational Materials Science, 2012, 59: 87-93.
[14] FAZELI F, SINCLAIR C W, BASTOW T. The role of excess vacancies on precipitation kinetics in an Al-Mg-Sc alloy[J]. Metallurgical and Materials Transactions A, 2008, 39(10): 2297-2305.
[15] WOODWARD C, ASTA M, KRESSE G, HAFNER J. Density of constitutional and thermal point defects in L12 Al3Sc[J]. Physical Review B, 2001, 63(9): 094103.
[16] KRESSE G, FURTHMULLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169-11186.
[17] CEPLERLEY D M, ALDER B J. Exchange-correlation potential for LDA[J]. Physical Review Letters, 1980, 45(7): 566-569.
[18] PERDEW J P, CHEVARY J A, VOSKO S H, JACKSON K A, PEDERSON M R, SINGH D J, FIOLHAIS C. Atoms, molecules, solids and surfaces: Applications of the generalized gradient approximation for exchange and correlation[J]. Physical Review B, 1992, 46(11): 6671-6687.
[19] PERDEW J P, BURKE K M, EMZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[20] KRESSE G, JOUBERT J. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59(3): 1758-1775.
[21] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192.
[22] BLOCHL P E, JEPSEN O, ANDERSEN O K. Improved tetrahedron method for Brillouin-zone integrations[J]. Physical Review B, 1994, 49(23): 16223-16233.
[23] CHETTY N, WEINERT M, RAHMAN T S, DAVENPORT J W. Vacancies and impurities in aluminum and magnesium[J]. Physical Review B, 1995, 52(9): 6313-6326.
[24] FISCHER T H, ALMLOF J. General methods for geometry and wave function optimization[J]. The Journal of Physical Chemistry, 1992, 96(24): 9768-9774.
[25] PULAY P. Ab inito calculation of force constants and equilibrium geometries in polyatomic molecules ��: Theory[J]. Molecular Physics, 1969, 17(2): 197-204.
[26] MURNAGHAN F D. The compressibility of media under extreme pressures[J]. Proceedings of the National Academy of Sciences of the United States of America, 1944, 30(9): 244-247.
[27] SEITZ F, TURNBUU D. Solid state physics: Advance in research and applications (Volume 16)[M]. New York: Academic Press, 1964.
[28]  M, HAFNER J. Response of trialuminides to [110] uniaxial loading: An ab initio study for Al3(Sc,Ti,V)[J]. Physical Review B, 2007, 76(1): 014110.
M, HAFNER J. Response of trialuminides to [110] uniaxial loading: An ab initio study for Al3(Sc,Ti,V)[J]. Physical Review B, 2007, 76(1): 014110.
[29] ��С��. ϡ������þ�Ͻ�����ѧ���ʵĵ�һԭ������[D]. ��ɳ: ���ϴ�ѧ, 2008.
TAO Xiao-ma. First-principles calculations of the thermodynamic properties of rare earths-aluminum and rare earths-magnesium alloys[D]. Changsha: Central South University, 2008.
[30] ASIA M, FOILES S M, QUONG A A. First-principles calculations of bulk and interfacial thermodynamic properties for fcc-based A1-Sc alloys[J]. Physical Review B, 1998, 57(18): 11265-11275.
[31] CACCIAMANI G, RIANI P, BORZONE G. Thermodynamic measurements and assessment of the Al-Sc system[J]. Intermetallics, 1999, 7(1): 101-108.
[32] �����, �� ��, �� ��, ������. Al-Sc �����仯����ĵ��ӽṹ���ȶ��Ժ�����ѧ����[J]. �й���ɫ����ѧ��, 2010, 20(5): 946-953.
LI Yan-feng, XU Hui, ZHANG Biao, ZHANG Li-gang. Electronic structure, stability and thermodynamic properties of Al-Sc intermetallics compounds[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(5): 946-953.
[33] ��˳ƽ, ��Сƽ, ¬����, �� ��, �Ƶ�Զ, ����. ����Miedemaģ��Al3X(Sc, Er, Zr, Li)��ȱ���γ��ʵļ���[J]. ϡ�н��������빤��, 2013, 42(7): 1478-1482.
SUN Shun-ping, LI Xiao-ping, LU Ya-lin, LI Yong, HUANG Dao-yuan, YI Dan-qing. Calculation of formation enthalpies of point defects for Al3X(Sc, Er, Zr, Li) intermetallics based on Miedema��s model[J]. Rare Metal Materails and Engineering, 2013, 42(7): 1478-1482.
[34] SUN S N, KIOUSSIS N, LIM S, GONIS A, GOURDIN W H. Impurity effects on atomic bonding in Ni3Al[J]. Physical Review B, 1995, 52(20): 14421-14430.
(�༭ ����)
������Ŀ��������Ȼ��ѧ����������Ŀ(51071177)������ʡ�ص�������Ŀ(11070203010)����������ѧԺ��ʿ������������(KYY12008)
�ո����ڣ�2012-09-28�������ڣ�2013-04-25
ͨ�����ߣ��� �£����ڣ���ʿ���绰��0731-88830450��E-mail: yjiang@csu.edu.cn

