文章编号:1004-0609(2009)08-1517-07
铁杂质对闪锌矿表面电子结构及活化影响的
第一性原理研究
陈建华1,陈 晔2,曾小钦2,李玉琼2
(1. 广西大学 资源与冶金学院,南宁530004;
2. 广西大学 化学化工学院,南宁 530004)
摘 要:采用基于密度泛函理论的平面波赝势法对含铁杂质闪锌矿(110)表面电子结构及铜在闪锌矿表面的吸附进行模拟。计算结果表明:铁杂质与锌原子的替换能均为负值,说明铁很容易替换闪锌表面的锌,且该替换反应对位置不敏感;铁杂质的存在导致表面能带向溶部移动,且在禁带中出现由铁3d轨道的tg和e2g构成的杂质能级,以及硫3p轨道分裂形成的Tamm表面态能级;铁杂质还改变了闪锌矿表面d态和sp态电子的分布;铁杂质的存在有利于闪锌矿表面锌原子与铜发生交换反应,但闪锌矿表面的铁不能与铜发生置换反应,从而减小闪锌矿表面与铜发生交换的总锌位,降低铜对闪锌矿的活化效果。
关键词:闪锌矿;铁杂质;密度泛出理论;铜活化
中图分类号:TD 923.13 文献标识码:A
First principle study of effect of Fe impurity on electronic structure and activation of sphalerite surface
CHEN Jian-hua1, CHEN Ye2, ZENG Xiao-qin2, LI Yu-qiong2
1. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;
2. College of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China
Abstract: The simulation of Fe doped sphalerite (110) surface and Cu adsorption on the sphalerite surface were performed by using plane wave-pseudopotential approach based on density-functional theory. The results show that the substitution energy for Fe substituted Zn atom are negative which indicate the Fe is easy to substitute Zn atom on sphalerite surface, and substitute reaction is insensitive to the site. The presence of Fe impurity shifts the energy band to deep part. Impurity level composed of tg and e2g of Fe 3d orbital and Tamm surface cased by the splitting of S 3p orbital occur in the band gap. Fe impurity changes the distribution of d state and sp state electrons. The calculation suggests that Fe doping sphalerite (110) surface is favorable for the exchange of Cu with Zn at the first layer, however, the Fe atom on the surface can not be replaced by Cu atom, which reduces the total Zn activation sites of sphalerite (110) surface, and the activation of copper on the sphalerite surface becomes poor.
Key words: sphalerite; iron impurity; density functional theory (DFT); copper activation
闪锌矿是一种主要的含锌矿物,且常与黄铁矿和方铅矿等硫化矿共生。闪锌矿一般采用浮选法进行回收,但闪锌矿的天然可浮性较差,直接采用黄药难以进行捕收,需要加入硫酸铜活化后才能浮选。天然闪锌矿常含有铁杂质,其含量受成矿条件的影响,铁杂质的含量可为0.4%~22%,含铁超过6.0%的闪锌矿称为铁闪锌矿[1]。铁闪锌矿是一种常见的硫化锌矿物,且是稀有金属铟的主要赋存矿物,是铟资源的主要来源。铁杂质对闪锌矿的可浮性具有较大影响,随着闪锌矿中含铁量的增高,闪锌矿越难活化,同时对石灰也越敏感。CHANDRA和GERSON[2]研究表明,铁杂质的存在降低了闪锌矿表面铜离子的吸附量[2]。BOULTON等[3]认为,闪锌矿晶格中存在的铁会减少铜离子与锌的交换位。但HARMER等[4]的研究却表明,闪锌矿中的铁含量增加了铜在表面的吸附量,与低铁含量的闪锌矿相比,铁含量的提高增加了表面缺陷的位置,使得更多的铜离子吸附到表面。
铁杂质对闪锌矿的半导体性质影响较大,天然闪锌矿是绝缘体,含铁闪锌矿则是n型半导体,铁含量越高,其导电性越强[4?5]。而半导体性质的改变会影响矿物表面电子结构以及药剂分子的吸附,从而改变硫化矿物的可浮性[6]。基于密度泛函理论的第一性原理是研究硫化矿物半导体晶格缺陷的有效方法,对黄铁矿、白铁矿、硫化锌和方铅矿等进行了晶格缺陷的计算。研究结果表明,晶格缺陷导致了硫化矿物能带结构、电子态密度以及原子电荷的变化[7?12]。而采用基于密度泛函理论的第一性原理研究晶格缺陷对硫化矿浮选的影响还未见报道。为了进一步从本质上了解铁杂质对闪锌矿浮选的影响,本文作者采用基于第一性原理的量子力学方法研究了铁杂质对闪锌矿(110)表面结构与电子性质的影响,同时考察铁杂质对铜活化闪锌矿的影响。
1 计算方法与模型
本计算采用CASTEP软件包完成,CASTEP采用的方法是密度泛函理论框架下的第一性原理[13]。计算中交换关联函数采用广义梯度近似(GGA)下的PBE梯度修正函数,采用超软赝势(Ultrosoft)描述离子实和价电子的相互作用[14]。各原子的赝势计算选取的价电子分别为Zn3d104s2、S3s23p4和Fe3d64s2,在优化掺铁原子体系的几何构型中考虑了自旋极化。平面波截断能设为310 eV,系统总能量和电荷密度在Brillouin区的积分计算采用Monkhorst-Pack方案来选择K网格点为2×3×1,以保证体系能量和构型在准完备平面波基水平上的收敛。在自洽场运算中,采用了Pulay密度混合法,自洽场收敛精度设为2.0×10?6 eV/atom。在对模型的结构优化中采用BFGS算法,参数优化如下:原子间相互作用力的收敛标准设为0.05 eV/?,晶体内应力的收敛标准设为0.1 GPa,原子最大位移收敛标准设为2×10?3 ?。对这3个参数同时进行优化,结构优化完成的标志是这些参数均达到收敛标准,计算结果令人满意。
闪锌矿ZnS有闪锌矿结构和纤维矿结构,本模拟取闪锌矿结构进行计算,其空间群为 ,属于等轴晶系,体对角线的1/4处为硫原子,8个角和6个面心处为锌原子,每个晶胞内含有4个Zn原子和4个S原子,晶格常数a=b=c=0.540 93 nm,α=β=γ=90?。国内外对闪锌矿的各种表面进行了研究,其中DUKE和WANG等[15]的研究表明,闪锌矿的(110)面的弛豫会导致较低的表面自由能,而WRIGHT等[16]的研究表明,闪锌矿(111),(
,属于等轴晶系,体对角线的1/4处为硫原子,8个角和6个面心处为锌原子,每个晶胞内含有4个Zn原子和4个S原子,晶格常数a=b=c=0.540 93 nm,α=β=γ=90?。国内外对闪锌矿的各种表面进行了研究,其中DUKE和WANG等[15]的研究表明,闪锌矿的(110)面的弛豫会导致较低的表面自由能,而WRIGHT等[16]的研究表明,闪锌矿(111),( )和(001)面的重构也会降低表面能,但这些面的表面能仍高于(110)的面,因此,选取闪锌矿(110)面作为计算对象。构建闪锌矿(110)面模型时,首先优化原胞,优化后的晶胞参数为0.545 51 nm,与实验值0.540 93 nm符合得较好。在优化后的原胞基础上构建出1个5层原子层的(110)面,真空层厚度为15 ?,其模型如图1所示。最后对构建好的(110)面进行几何优化,进行优化时固定下面三层,弛豫表面两层原子。构建含铁杂质的闪锌矿(110)面模型时,将一个铁原子替代(110)表面中的一个锌原子,掺铁原子浓度为1.25%。在表面每层中有两个可能的杂质位置,分别对这6个杂质位置进行几何优化,并且定义在闪锌矿(110)表面一个原子X(Fe 或Cu)替换一个原子M(Zn或Fe )的置换能计算公式如下:
)和(001)面的重构也会降低表面能,但这些面的表面能仍高于(110)的面,因此,选取闪锌矿(110)面作为计算对象。构建闪锌矿(110)面模型时,首先优化原胞,优化后的晶胞参数为0.545 51 nm,与实验值0.540 93 nm符合得较好。在优化后的原胞基础上构建出1个5层原子层的(110)面,真空层厚度为15 ?,其模型如图1所示。最后对构建好的(110)面进行几何优化,进行优化时固定下面三层,弛豫表面两层原子。构建含铁杂质的闪锌矿(110)面模型时,将一个铁原子替代(110)表面中的一个锌原子,掺铁原子浓度为1.25%。在表面每层中有两个可能的杂质位置,分别对这6个杂质位置进行几何优化,并且定义在闪锌矿(110)表面一个原子X(Fe 或Cu)替换一个原子M(Zn或Fe )的置换能计算公式如下:

?Es越负说明置换反应越容易进行。
2 结果与讨论
2.1 几何优化与表面弛豫
从图1所示模型可知,闪锌矿(110)面每一层有两种不同的锌位置,分别定义为顶位(T位)和底位(B位),因此,铁杂质在每一层与锌的替换都存在两种情况,采用第一性原理对铁原子替代闪锌矿(110)表面锌原子的可能位置进行了几何优化。表1所列为铁替代不同位置上的锌的能量。
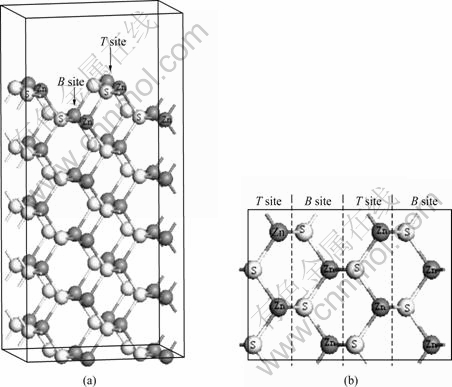
图1 闪锌矿(110)面的模型示意图
Fig.1 Schematic diagrams of model of sphalerite (110) surface: (a) Side view; (b) Top view
表1 铁杂质替换不同位置锌的替换能
Table 1 Substitution energy ?Es for Fe impurity substitute different sites of Zn
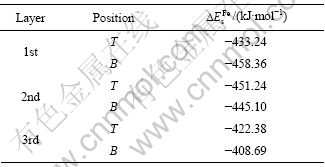
从表1可知,铁原子替代锌原子的 值较负,说明铁原子很容易代替闪锌矿晶格中的锌原子,这也是铁闪锌矿在自然界中广泛存在的原因。另外,铁在第一层的B位和第二层的T位和B位的替换能比较接近,说明铁原子与这3个位置的锌原子置换较容易发生。由于浮选是在矿物表面最外层进行的,因此,本文作者重点讨论表面第一层B位的铁杂质对闪锌矿(110)表面结构和性质的影响。
值较负,说明铁原子很容易代替闪锌矿晶格中的锌原子,这也是铁闪锌矿在自然界中广泛存在的原因。另外,铁在第一层的B位和第二层的T位和B位的替换能比较接近,说明铁原子与这3个位置的锌原子置换较容易发生。由于浮选是在矿物表面最外层进行的,因此,本文作者重点讨论表面第一层B位的铁杂质对闪锌矿(110)表面结构和性质的影响。
对于理想的和含铁杂质的闪锌矿(110)面表面两层进行弛豫,定义沿坐标轴正方向的位移为正,沿坐标轴负方向的位移为负。分析弛豫结果表明,理想的闪锌矿(110)表面原子位置在z轴方向变化很小,说明表面结构变化不大。另外,当闪锌矿(110)面含铁杂质时,表面原子弛豫主要是与铁原子相连的两个硫原子,硫原子在z轴方向向下移动0.42%,同时在x轴方向产生约3%的弛豫。说明铁杂质对于与其相邻的硫原子影响较大,所引起的变形也较显著。
2.2 能带结构与态密度
理想和含铁杂质的闪锌矿(110)面能带结构如图2所示。由图2(a)可以看出,理想闪锌矿(110)面计算所得的禁带宽度为2.13 eV,小于试验值3.72 eV,这个差异是由于GGA近似下的DFT对电子与电子之间的交换关联作用处理不足引起的,但不影响对结果的分析。由图2(b)可知,闪锌矿(110)表面掺铁导致禁带宽度变窄,且能带向深部移动,说明体系电子数增加。另外掺铁后,费密能级更靠近导带,表明掺铁后闪锌矿表面由p型半导体变成n 型。另外,从图2还可以明显看出,掺铁后闪锌矿矿(110)面禁带中?0.65 eV和0.04 eV附近出现表面能级。结合态密度分析可知,禁带中的表面态能级由两部分构成,一部分是由于铁在四面体场中发生分裂形成的杂质能级;另一部分是由于铁杂质存在,导致硫3p轨道发生分裂形成的表面态能级,即Tamm表面态。
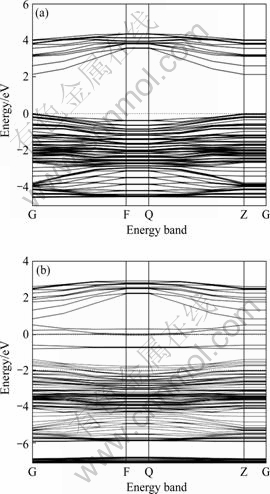
图2 闪锌矿(110)面的能带结构
Fig.2 Band structures of ZnS(110): (a) Perfect ZnS(110) surface; (b) Fe doped ZnS(110) surface
理想闪锌矿(110)表面原子的总电子态密度以及Zn 4s, Zn 3d, S 3s 和 S 3p的分态密度分别如图3所示。从图3可以看出,在价带深部 -11.75 eV出现的峰值主要是由硫原子的S 3s轨道构成。在价带上部-5.85 eV出现的峰值主要是由锌原子的Zn 3d和硫原子的S 3p轨道组成。导带主要是由锌原子的Zn 3s轨道和硫原子的S 3p轨道组成。
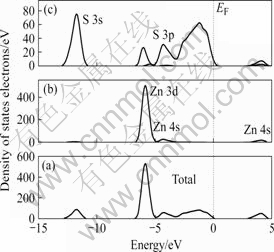
图3 理想闪锌矿(110)面的态密度和分态密度图
Fig.3 DOS and PDOS plots of perfect ZnS(110)
含铁杂质的闪锌矿(110)表面原子的总电子态密度和Zn 4s、Zn 3d、S 3s、S 3p 以及 Fe 3d的分态密度分别如图4所示。由图4可见,在价带深部由硫原子S 3s轨道构成的峰从 -11.75 eV移动到-13.28 eV,另外硫3p轨道也有较大幅度的负移,且表面层的S 3p轨道在费密能级附近形成了一个新的峰。这主要是由于铁原子的电负性(1.85)比锌原子的(1.60)大,因此,铁原子对硫原子电子的吸引力比锌原子大,从而导致S 3s 和S 3p电荷的减少,能带向深部移动,Mulliken电荷布居分析也证实了这一现象。价带中锌原子的 Zn 3d轨道从 -5.85eV 移动到 -7.37 eV,这主要是由于表面缺少1个锌原子的Zn 3d轨道的贡献,从而导致偏移。而铁杂质的影响主要在费米能级附近。Fe 3d轨道在费密能级附近发生了分裂,形成t2g (-0.65 eV)和eg (0.04 eV)两个峰。

图4 含铁闪锌矿(110)面的态密度和分态密度
Fig.4 DOS and PDOS plots of Fe doped ZnS(110) surface
2.3 表面电荷分析
跟浮选药剂作用密切相关的是矿物表面最外层的原子,理想以及含铁闪锌矿(110)表面第一层的sp态和d态电子分布如表2所列。从表2中可知,对于理想闪锌矿,与闪锌矿体相的sp电子数(64)和d电子数(80)相比,表面顶层d电子数减少至79.8,而sp电子数增加到65.4。这是由于表面之外没有原子,从而引起电子分布适当调整,顶层电子数增加,在表面形成偶极层和相应的自洽表面势,使顶层的d电子略微减少,sp电子数增多[17]。
当闪锌矿表面含有铁杂质时,与理想闪锌矿表面电子分布变化相反,第一层d电子数增加0.76,而sp电子则减少0.54,表明表面铁杂质的存在能够补偿周期性势场截断而引起自洽表面势,从而改变了表面d态和sp态电子的分布。
表2 铁原子对闪锌矿(110)表面第一层原子的sp态和d态电子分布的影响
Table 2 Effect of Fe heteroatom on electron distribution of sp and d states at 1st layer of sphalerite (110) surface
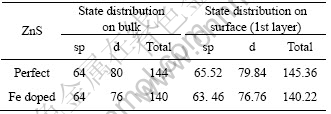
图5所示为理想闪锌矿及含铁闪锌矿(110)表面第一层各原子的Mulliken电荷。由图1(b)可知,闪锌矿(110)表面原子存在两种位置:顶位(T位)和底位(B位)。从图5(a)可见,这两种位置上的锌原子和硫原子的Mulliken电荷数是不同的,T位的锌原子和硫原子的Mulliken电荷数分别为0.35和?0.50,而B位的锌原子和硫原子的Mulliken电荷数分别为0.31和?0.49。当掺杂铁原子后,闪锌矿表面的Mulliken电荷数重 新分布。由图5(b)可见,铁原子的Mulliken电荷数为0.12,比锌原子低,这是因为铁原子的电负性比锌原子的电负性大,所以铁原子吸引电子的能力更强,从而导致铁原子比锌原子电荷数小。图6所示为理想的及含铁闪锌矿(110)表面差分电荷密度。比较图6(a)和(b)所示的电荷差分密度图也可以看出,与铁相连的硫原子的电子云明显向铁原子偏移。从图5(b)可知,与铁相连的硫原子的Mulliken电荷数从?0.49减少到?0.31左右,另外与铁相邻的锌原子的电荷数也发生了变化。以上结果表明,铁原子对闪锌矿表面电子结构及电荷分布有较大影响,从而影响闪锌矿的可浮性。

图5 理想的及含铁闪锌矿(110)面的Mulliken电荷
Fig.5 Mulliken charge of perfect (a) and Fe doped (b) sphalerite (110)

图6 理想的及含铁闪锌矿(110)表面差分电荷密度图
Fig.6 Contour plots of electron density difference for perfect (a) and Fe doped (b) sphalerite (110) surface
2.4 铁杂质对铜离子活化闪锌矿的影响
铜离子对闪锌矿的活化是闪锌矿能否浮选的一个必要条件,国内外对闪锌矿的活化机理有很多研究,结果表明硫酸铜活化闪锌矿是一个离子交换过程,Cu(Ⅱ)在闪锌矿表面的吸附会释放出等量的Zn2+到溶液中,反应式如下[2]:
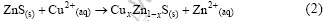
SIMS和XPS分析结果表明[19],在低pH值下,到达闪锌矿表面的Cu2+离子立刻被还原成Cu+离子,且与表面任意位置的锌发生替换,从而将锌离子释放到溶液中。当铜离子浓度比较高时,在闪锌矿表面第二层及其它层将会发生类似的Cu+离子与Zn2+离子的替换反应,而这种反应可能是通过表面层的Cu+离子与第二层的锌的交换或者是表面的Cu2+离子通过间隙进入闪锌矿的次表面层而发生交换反应。
关于铁杂质对铜活化闪锌矿的影响仍是有争议的,为了进一步理解铜活化闪锌矿表面的机理,弄清铁杂质对闪锌矿吸附铜离子的影响,采用第一性原理对闪锌矿表面、次表面和第三层的不同位置的锌和铜交换进行了优化计算。表3所列为铜替换不同位置锌的替换能。由表3可知,铜与闪锌矿表面层T位的替换能为?82.09 kJ/mol,是所有替换能中最低的位置,说明铜与硫化锌(110)表面锌原子发生替换反应的稳定位置是第一层的T位。
表3 铜替换不同位置锌的替换能
Table 3 Substitution energy ( ) of possible Zn sites replaced by Cu
) of possible Zn sites replaced by Cu
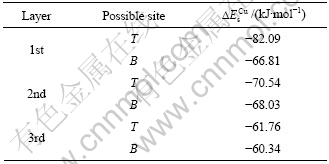
当闪锌矿表面存在铁杂质的时候,需要考虑如下两个问题:一是表面的铁能否被铜替换,二是铁杂质对表面锌与铜替换的影响。根据前面的计算结果可知,铁杂质在闪锌矿表面第一层的B位和第二层的T位、B位都有可能存在,因此,根据式(3)计算出铜与闪锌矿(110)表面这3个位置上的铁杂质的替换能?Es分别为391.21、380.35和376.73 kJ/mol,由于?Es为正值,说明式(3)的置换反应不能发生,即闪锌矿表面和次表面的Fe杂质均不能被Cu替换出来。
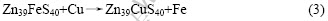
其次考察闪锌矿表面的Fe杂质对表面锌与铜替换的影响。根据式(4)的反应计算出铜与含铁闪锌矿(110)表面的锌原子的替换能?Es为?194 kJ/mol,比闪锌矿(110)面锌原子与铜发生替换反应的替换能?82.09 kJ/mol更低,说明式(4)比式(2)更容易发生,即闪锌矿表面铁杂质的存在有利于铜与锌原子发生置换反应。这可能是由于铁杂质的存在导致了表面锌原子位置较大的弛豫,另外由于铁原子的电负性比锌原子大,导致表面层的硫原子电子云向铁原子方向偏移,从而降低了硫原子对锌原子的束缚。
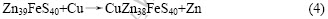
以上结果表明,表面铁杂质有利于闪锌矿(110)面锌与铜进行离子交换吸附,但表面的铁杂质不能被铜置换,减少了闪锌矿表面活化点数量,从而降低了铜在铁闪锌矿表面的总吸附量。可以预测闪锌矿中铁含量越大,越不利于铜对闪锌矿表面的活化。这与铁闪锌矿的浮选实践现象和分析结果一致。
3 结论
1) 闪锌矿(110)表面掺铁导致能带向深部移动,掺铁闪锌矿(110)电子构型更稳定。掺铁后闪锌矿表面由p型半导体变成n型。另外,掺铁后闪锌矿(110)面禁带出现了表面能级,这是由于铁的3d轨道和硫3p轨道分裂形成的杂质能级和Tamm表面态能级。
2) 闪锌矿表面铁杂质的存在能够补偿周期性势场截断而引起自洽表面势,从而改变了表面d态和sp态电子的分布。与理想闪锌矿表面电子分布变化相反,掺铁闪锌矿(110)面第一层d电子数增加了0.76,而sp电子则减少了0.54。
3) 由于铁原子的电负性比锌原子的电负性大,导致与铁相连的硫原子的电子云明显向铁原子偏移,硫原子的Mulliken电荷数从?0.49减少到?0.31左右,铁硫键的共价性增强。
4) 表面铁杂质有利于闪锌矿(110)面锌与铜进行离子交换吸附,但表面的铁杂质不能与铜发生交换,导致闪锌矿表面活化点数量减少,从而降低了铜在铁闪锌矿表面的吸附量。且闪锌矿中铁含量越大,越不利于铜对闪锌矿表面的活化。
REFERENCES
[1] TONG Xiong, SONG Shao-xian, HE Jian. Activation of high-iron marmatite in froth flotation by ammoniacal copper(II) solution[J]. Miner Eng, 2007, 20(9): 259?263.
[2] CHANDRA A P, GERSON A R. A review of the fundamental studies of the copper activation mechanisms for selective flotation of the sulfide minerals, sphalerite and pyrite[J]. Adv Colloid Interface Sci, 2009, 145(1): 97?110.
[3] BOULTON A, FORNASIERO D, RALSTON J. Effect of iron content in sphalerite on flotation[J]. Miner Eng, 2005, 18(9): 1120?1122.
[4] HARMER S L, MIERCZYNSKA V A, BEATTIE D A. The effect of bulk iron concentration and heterogeneities on the copper activation of sphalerite[J]. Miner Eng, 2008, 21(11): 1005?1012.
[5] 熊小勇. 铁成分对硫化锌精矿的半导体性质及化学反应性的影响[J]. 有色金属, 1989, 41(4): 55?66.XIONG Xiao-yong. Effect of the iron content of zinc sulphide concentrates on their semi-conductivity and chemical reactivity[J]. Nonferrous Metal, 1989, 41(4): 55?66.
[6] 陈建华, 冯其明, 卢毅屏. 电化学调控浮选能带理论及应 用(I)―半导体?溶液界面能带理论及模型[J]. 中国有色金属学报, 2000, 10(2): 240?242.CHEN Jian-hua, FENG Qi-ming, LU Yi-ping. Energy band model of electrochemical flotation and its application(Ⅰ)―Theory and model of energy band of semiconductor-solution interface[J]. The Chinese Journal of Nonferrous Metals, 2000, 10(2): 240?242.
[7] VON OERTZEN G U, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. J Electron Spectrosc, 2005, 144/147(6): 1245?1247.
[8] MARTIN R, UDO B. First-principle calculations of the thermodynamic mixing properties of arsenic incorporation into pyrite and marcasite[J]. Chem Geol, 2006, 225(3/4): 278?290.
[9] MARC B, MARIA A, JOHN B, KATE W, RICHARD C, CATLOW A. Arsenic incorporation into FeS2 pyrite and its influence on dissolution: a DFT study[J]. Geochim Cosmochim Ac, 2007, 71(3): 624?630.
[10] 何开华, 余 飞, 姬广富, 颜其礼, 郑澍奎. 第一性原理研 究ZnS掺V的光学性质和电子结构[J]. 高压物理学报, 2006, 20(1): 56?59.HE Kai-hua, YU Fei, JI Guang-fu, YAN Qi-li, ZHENG Shu-kui. Study of optical properties and electronic structure of V in ZnS by first principles[J]. Chinese Journal of High Pressure Physics, 2006, 20(1): 56?59.
[11] 肖 奇, 邱冠周, 胡岳华. 黄铁矿机械化学的计算模拟(Ⅰ)―晶格畸变与化学反应活性的关系[J]. 中国有色金属学报, 2001, 11(5): 900?904.XIAO Qi, QIU Guan-zhou, HU Yue-hua. Computational simulation to mechanical activation of pyrite(I)―Relation of structural strain to chemistry reaction activity[J]. The Chinese Journal of Nonferrous Metals, 2001, 11(5): 900?904.
[12] 丁宗玲, 郭 英, 邢怀中, 李 伟, 陈效双. PbS电子结构的第一性原理[J]. 东华大学学报, 2008, 34(4): 512?516.DING Zong-ling, GUO Ying, XING Huai-zhong, LI Wei, CHEN Xiao-shuang. The First principle of the electronic structure of PbS[J]. Journal of Donghua University, 2008, 34(4): 512?516.
[13] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Rev Mod Phys, 1989, 61(3): 689?746.
[14] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phy Rev Lett, 1996, 77(18): 3865?6868.
[15] DUKE C B, WANG Y R. Surface structure and bonding of cleavage of tetrahedrally coordinated Ⅱ-Ⅳ compounds[J]. J Vac Sci Technol B, 1988, 6: 1440?1443.
[16] WRIGHT K, WATSON G W, PARKER S C, VAUGHAN D J. Simulation of the structure and stability of sphalerite (ZnS) surfaces[J]. Ameri Mineralogist, 1998, 83: 141?146.
[17] KAZUME N, MASAHITO Y, MASAYUKI H. Energetics of Mg and B adsorption on polar zinc oxide surfaces from first principles[J]. Phys Rev B, 2008, 77(3): 035330-1?6.
[18] SAWATZKY G A, ALLEN J W. Magnitude and origin of the band gap in NiO[J]. Phys Rev Lett, 1984, 53: 2339?2342.
[19] GERSON A R, LANGE A G, PRINCE E K, The mechanism of copper activation of sphalerite[J]. J Appl Surf Sci, 1999, 137(1/4): 207?223.
基金项目:国家自然科学基金资助项目(50684001)
收稿日期:2009-02-16;修订日期:2009-04-24
通讯作者:陈 晔,博士;电话:0771-3233566;E-mail: fby18@126.com
(编辑 龙怀中)

