文章编号:1004-0609(2010)04-0765-07
含空位和杂质缺陷的闪锌矿电子结构的第一性原理计算
陈建华1,曾小钦2,陈 晔1,张辉鹏2
(1. 广西大学 资源与冶金学院,南宁 530004;2. 广西大学 化学化工学院,南宁 530004)
摘 要:采用基于密度泛函理论(DFT)的平面波超软赝势方法,研究硫空位、锌空位以及铁和镉杂质对闪锌矿电子结构的影响,分析空位和杂质对闪锌矿的价键结构、能带、态密度、差分电荷密度等的影响。计算结果表明:镉杂质缺陷导致闪锌矿的晶胞参数变大,而硫空位、锌空位和铁杂质均使闪锌矿的晶胞参数变小;硫空位使闪锌矿的带隙变窄,与硫空位相邻的4个锌原子的电荷明显低于其他锌原子的电荷;锌空位使闪锌矿的带隙变宽,费米能级向低能方向偏移,与锌空位相邻的4个硫原子的电荷明显低于其他硫原子的电荷;铁杂质使闪锌矿的带隙变宽,并在带隙中形成一个由铁的3d轨道贡献的杂质能级,费米能级向高能方向偏移;镉杂质对闪锌矿能带结构和态密度的影响较小,在闪锌矿价带-7.5 eV处形成一个由镉的4d轨道贡献的能级,Cd―S键布居数下降,共价性减弱。
关键词:闪锌矿;空位缺陷;铁杂质;镉杂质;电子结构;第一性原理
中图分类号:TD923.13 文献标志码:A
First-principle theory calculations of electronic structure of
sphalerite with vacancy and impurity
CHEN Jian-hua1, ZENG Xiao-qin2, CHEN Ye1, ZHANG Hui-peng2
(1. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;
2. College of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China)
Abstract: The electronic structures of sphalerite with Zn-vacancy, S-vacancy, Fe-impurity and Cd-impurity were calculated, respectively, using the ultra-soft pseudo-potential approach of the plane wave based on the density functional theory(DFT), and their bond structure, band structure, density of states and the difference charge density were studied. The calculated results indicate that the Cd-impurity makes the lattice constant of sphalerite increase, but the S-vacancy, Zn-vacancy and Fe-impurity make the lattice constant decrease. The S-vacancy makes the band gap become narrow, and the Mulliken charge of Zn-atoms around the S-vacancy is lower than that of others. On the contrary, the Zn-vacancy makes the band gap become wide and the Fermi level moves to a low energy level, and the Mulliken charges of S-atoms around Zn-vacancy are lower than those of others. The Fe-impurity makes the band gap become wide and forms an impurity level in band gap,which consists of the 3d-electrons of the Fe atom, and the Fermi level moves to a high energy level. The Cd-impurity has a little effect on the band structure and density of states of sphalerite, and there is a level in -7.5 eV formed by the 4d-electron of the Cd atom. The population of Cd―S bond decreases and the covalence becomes weak.
Key words: sphalerite; vacancy defect; iron-impurity; cadmium-impurity; electronic structure; first principle theory
闪锌矿属等轴晶系,硫离子成紧密堆积,锌离子位于半数四面体空隙中,配位数为4。它的化学组分为67.10%Zn和32.90%S。最常见的类质同象混入物为铁,其次为锰、镉、铟、镓、汞、锗等。此外,闪锌矿中还含有铜、锡、锑、铋等杂质。在硫化矿浮选实践中,人们发现不同矿床或同一矿床不同区段的同一种矿物,其浮选行为存在很大差异。对于闪锌矿,人们也发现不同矿床或同一矿床不同矿段的闪锌矿由于杂质不同而具有不同的颜色,从浅绿色、棕褐色和深褐色直到钢灰色。各种颜色的闪锌矿可浮性差别比较大,含镉的闪锌矿可浮选性比较好,而含铁的闪锌矿可浮性较差。硫化矿物浮选行为与矿物的半导体性质密切相关[1]。研究人员对黄铁矿、白铁矿、硫化锌、方铅矿等进行了晶格缺陷的计算,研究结果:表明晶格缺陷导致硫化矿物能带结构、电子态密度以及原子电荷的变化[2-12]。硫化矿物的半导体性质与矿物晶格缺陷及缺陷浓度有密切关系,不同产地的硫化矿物由于成矿条件和环境的不同,其矿物晶胞参数和缺陷也不同,从而导致不同产地同一种矿物浮选行为的差异。
在浮选实践中,经常碰到的闪锌矿缺陷主要有两大类:一是化学计量系数的偏离,即锌硫比偏离1?1,这主要是由空位缺陷造成的;二是铁和镉杂质,即闪锌矿晶格中含铁或者镉杂质。天然闪锌矿常含有铁杂质,其含量(质量分数)从0.4%到22%不等,含铁超过6.0%的闪锌矿称为铁闪锌矿[13]。铁杂质对闪锌矿的半导体性质影响较大,含铁闪锌矿是n型半导体,铁含量越高,其导电性越强[14-15],铁杂质使闪锌矿不利于或者难于浮选。闪锌矿中也常含有镉杂质,当含镉量达到5%时,称为镉闪锌矿,镉杂质能够提高闪锌矿的可浮性。本研究根据矿物晶体学和半导体缺陷理论构建含有硫空位、锌空位、铁杂质和镉杂质的闪锌矿模型,应用基于密度泛函理论的第一性原理,采用GGA-PBE交换关联势,计算和研究这两种空位和两种杂质对闪锌矿的几何结构、能带、态密度、差分电荷密度等电子结构的影响。研究结果对于进一步查清楚晶格缺陷对闪锌矿浮选行为影响的本质具有重要的学术意义和应用价值。
1 理论模型和计算方法
1.1 理论模型
闪锌矿的空间群为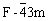 ,每个晶胞中包含4个锌原子和4个硫原子,在对角线的1/4处为硫原子,8个角和6个面心为锌原子。本研究主要考虑硫空位、锌空位、铁杂质和镉杂质4种常见的闪锌矿缺陷,并考虑实际矿物中的缺陷含量,构建出不同缺陷含量的晶胞模型。对于硫空位、锌空位和镉杂质,分别建立 2×1×1(Zn8S7,Zn7S8,Zn7CdS8)、2×2×1(Zn16S15, Zn15S16,Zn15CdS16)和2×2×2(Zn32S31,Zn31S32, Zn31CdS32)的超晶胞模型(见图1),缺陷的摩尔分数分别为6.25%、3.13%和1.56%,n(Zn)?n(S)在0.8~1.1之间;对于铁杂质,分别建立1×1×1(Zn3FeS4)、2×1×1(Zn7FeS8)和2×2×1(Zn15FeS16)的超晶胞模型(见图1),其中铁杂质的摩尔分数分别为12.50%、6.25% 和3.13%。
,每个晶胞中包含4个锌原子和4个硫原子,在对角线的1/4处为硫原子,8个角和6个面心为锌原子。本研究主要考虑硫空位、锌空位、铁杂质和镉杂质4种常见的闪锌矿缺陷,并考虑实际矿物中的缺陷含量,构建出不同缺陷含量的晶胞模型。对于硫空位、锌空位和镉杂质,分别建立 2×1×1(Zn8S7,Zn7S8,Zn7CdS8)、2×2×1(Zn16S15, Zn15S16,Zn15CdS16)和2×2×2(Zn32S31,Zn31S32, Zn31CdS32)的超晶胞模型(见图1),缺陷的摩尔分数分别为6.25%、3.13%和1.56%,n(Zn)?n(S)在0.8~1.1之间;对于铁杂质,分别建立1×1×1(Zn3FeS4)、2×1×1(Zn7FeS8)和2×2×1(Zn15FeS16)的超晶胞模型(见图1),其中铁杂质的摩尔分数分别为12.50%、6.25% 和3.13%。
1.2 计算方法
本研究所有的计算都采用CASTEP[16]软件来完成。在对模型进行几何优化和性质计算时,采用BFGS优化算法,交换关联函数采用广义梯度近似(GGA)[17]
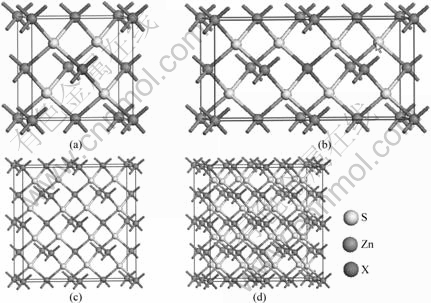
图1 4种不同缺陷超晶胞模型代表图
Fig.1 Four kinds of super-cell structures of ZnS with defect: (a) 1×1×1 super-cell; (b) 2×1×1 super-cell; (c) 2×2×1 super-cell; (d)2×2×2 super-cell(X= vacancy or impurity atom)
下的PBE梯度修正函数,采用超软赝势[18]描述价电子和离子间的相互作用。平面波截断能经过收敛测试后取330 eV,K点设置使用Monkhorst-Pack方案,铁杂质模型1×1×1、2×1×1和2×2×1的超晶胞分别采用4×4×4、2×4×4和2×2×4的K点网络,空位和镉杂质模型2×1×1、2×2×1和2×2×2的超晶胞分别采用2×4×4、2×2×4和2×2×2的K点网络,保证了体系能量和构型在准完备平面波基组水平上的收敛。收敛标准设置为:原子位移不大于0.000 2 nm,原子间作用力不大于0.05 eV/nm,原子间的内应力不大于0.1 GPa,体系总能量的变化不大于20 μeV/atom,所有的计算均在倒易空间进行。计算含铁杂质的超晶胞时还考虑了铁的电子自旋作用,参与计算的价态电子为S3s23p4、Zn3d104s2、Fe3d64s2和Cd4d105s2。
2 计算结果与讨论
2.1 闪锌矿的电子结构
首先,我们对ZnS原胞进行几何优化,然后,再对它的电子性质进行计算。通过计算得到ZnS晶格常数为0.542 7 nm,与试验值0.541 4 nm[19]接近,误差仅为0.24%。计算得到的ZnS带隙的宽度为2.18 eV,小于试验值3.72 eV[20]。这个差异是GGA或LDA近似下的DFT对电子与电子之间的交换关联作用处理不足引起的[21],不影响对能带和电子结构的分析。
从图2可以看出,理想ZnS的价带极大值(VBM)和导带极小值(CBM)都是位于高对称G点(Γ点) ,是一个直接带隙p型半导体。价带延伸至-13.57 eV,整个价带可以分为上、下两个部分,相对于价带,导带的变化则要平缓些。由图2(b)可见,位于-11.7 eV附近的下价带主要是由硫的3s轨道贡献,锌的4s轨道也贡献了一部分;在上价带中位于-5.9 eV的峰值主要是锌的3d轨道的贡献,其次是硫的3p轨道的贡献;价带的其他部分则主要是由硫的3p轨道和锌的4s轨道共同组成。导带主要由锌的4s轨道和硫的3p轨道共同组成。Mulliken布居分析表明:形成ZnS晶体时,主要由硫的3p轨道和锌的3d轨道来贡献,Zn―S键表现为共价键的性质,硫和锌原子所带电荷分别为-0.48e和0.48e。
2.2 缺陷对闪锌矿能带结构及态密度的影响
2.2.1 缺陷对闪锌矿几何结构的影响
对硫空位、锌空位、铁杂质和镉杂质的摩尔分数为3.13%的闪锌矿超晶胞进行结构优化。结果表明:
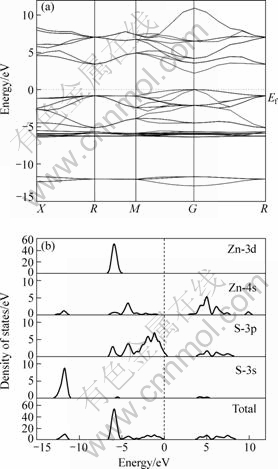
图2 ZnS的能带结构和分态密度图
Fig.2 Band structure (a) and partial density of states (b) of ZnS
由于硫空位、锌空位的存在,导致闪锌矿的晶胞参数变小,与理想闪锌矿的晶胞参数相比,含硫空位、锌空位的闪锌矿的晶胞参数分别减小了2.4%和1.3%。另外由于硫空位的存在,导致闪锌矿中空位周围的原子向空位中心偏移,特别是与硫空位相邻的4个锌原子偏移较明显;但是锌空位对闪锌矿超晶胞的几何结构没有明显影响,原子仅在空位周围驰豫。这是由于硫空位比锌空位体积大,导致硫空位周围的原子更容易变形。
对于杂质缺陷,优化结果表明含铁杂质闪锌矿的晶胞参数基本不变,而镉杂质导致闪锌矿晶胞参数变大。这是由于铁原子半径为0.124 nm,与锌原子半径0.132 nm相近,因此含铁杂质闪锌矿晶胞参数略有减小。而镉原子半径为0.148 nm,大于锌原子的半径,从而导致晶胞参数变大。
2.2.2 缺陷对闪锌矿能带结构的影响
图3所示分别为含有摩尔分数为3.13%的硫空位、锌空位、铁杂质和镉杂质的闪锌矿的能带结构。从图3可以看出:硫空位、锌空位和镉杂质不改变闪锌矿半导体类型,均为直接带隙p型半导体,但是含硫空位闪锌矿的价带最大值和导带最小值位于F点,而不是G点;铁杂质导致闪锌矿从p型半导体转变成n型半导体。从图3还可看出,在这4种缺陷中,镉杂质和硫空位降低了闪锌矿的带隙;而铁杂质和锌空位却增大了闪锌矿的带隙,铁杂质使费米能级向高能方向偏移,锌空位使费米能级向低能方向偏移,并在价带出现简并态。在计算中我们还考察了缺陷浓度对闪锌矿能带的影响,结果表明随着硫空位、镉杂质缺陷浓度的增大,闪锌矿的带隙都变窄,但硫空位和锌空位浓度对闪锌矿带隙变窄影响不显著。铁杂质在带隙中形成一个杂质能级,而镉杂质却在价带中的-7.5 eV处形成一个能级。
2.2.3 缺陷对闪锌矿态密度的影响
图4所示为含有摩尔分数为3.13%的硫空位、锌空位、铁杂质以及镉杂质的闪锌矿的总态密度图。从图4可见,硫空位导致位于-12.5 eV处的态密度峰值下降,这主要是由于闪锌矿超晶胞缺少一个硫原子,从而使3s轨道的贡献降低。而锌空位主要是影响位于-6.0 eV处的态密度峰值,这是因为晶胞中缺少一个锌原子,使锌的3d轨道在此处的贡献下降;另外,由于能带中-12.0 eV处的态密度包含了锌的4s轨道的贡献,因此锌空位也导致此处的态密度峰值下降。铁杂质在禁带中形成的杂质能级主要是铁的3d轨道的贡献,并且3d轨道在晶体场的作用下产生分裂,形成t2g和eg 两个新的杂质能级;由于体系中有一个锌原子被铁原子取代,导致主要由锌的3d轨道组成位于-7.2 eV处的态密度峰值下降。而镉杂质在价带-7.5 eV处形成的能级由镉的4d轨道来贡献。
2.3 缺陷闪锌矿的Mulliken布居分析
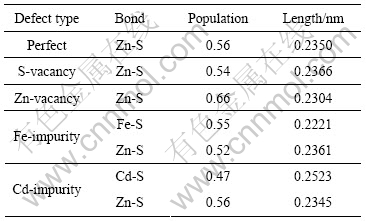
表1和2所列为含有摩尔分数为3.13%的硫空位、锌空位、铁杂质和镉杂质的闪锌矿中,与空位和杂质原子相邻的原子和键的Mulliken布居数。从表1和2可见:由于硫空位的影响,导致与硫空位相邻的4个锌原子的电荷为0.31e,明显低于0.48e,这主要是空位处缺少硫原子,减小了对锌原子的吸引作用,从而使锌原子失去的电子数减少。另外与硫空位相邻的Zn―S键的布居数减小,共价性减弱,键长变长。锌空位的存在导致与锌空位相邻的4个硫原子的电荷下降为-0.41e,这主要是缺少锌原子来贡献电子,导致硫原子所得电子数减少。另外,锌空位导致与其相邻的Zn―S键布居数增大,共价性增强,键长变短。由于铁原子的电负性比锌原子的强,不容易失去电子,导致与铁原子相连的硫原子的电荷下降为-0.40e。从表1和2可见:铁原子s和p轨道失去电子,而d轨道则得到电子,铁原子的Mulliken电荷为0.02e,且形成的Fe―S键布居数比相邻Zn―S键布居数大,共价性增强。这主要是铁原子的电负性比锌原子的大,导致电子云向铁原子偏移。由于镉与锌是同族元素,所以表现出与锌相似的Mulliken布居特征,形成的Cd―S键布居数下降,共价性减弱,键长变长;而与镉原子相邻的Zn―S键的布居数保持不变,但是键长变短。这主要是由于镉原子的半径比锌原子的大,使相邻硫原子向外扩张,导致Zn―S键的键长在一定范围内变短。
图3 含有硫空位、锌空位、铁杂质和镉杂质的闪锌矿的能带结构
Fig.3 Band structures of ZnS with S-vacancy (a), Zn-vacancy (b), Fe-impurity (c) and Cd-impurity (d)
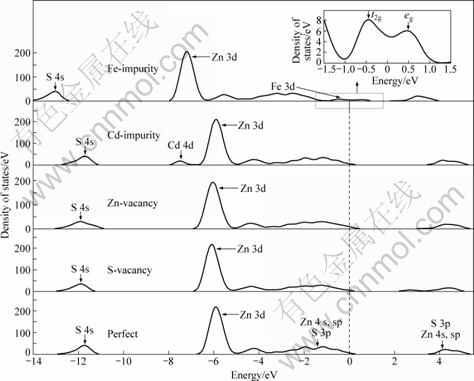
图4 含有硫空位、锌空位、铁杂质和镉杂质的闪锌矿的总态密度图
Fig.4 Total density of states of ZnS with S-vacancy, Zn-vacancy, Fe-impurity and Cd-impurity
表1 与硫空位、锌空位、铁杂质和镉杂质相邻的原子的Mulliken 电荷分析
Table 1 Mulliken atomic population analysis of atoms around S-vacancy, Zn-vacancy, Fe-impurity and Cd-impurity

表2 与硫空位、锌空位、铁杂质和镉杂质相邻的键布居分析
Table 2 Mulliken bond population analysis of bonds around S-vacancy, Zn-vacancy, Fe-impurity and Cd-impurity
2.4 缺陷闪锌矿的电荷密度和电荷差分密度分析
图5所示分别为含有摩尔分数为3.13%的硫空位、锌空位、铁杂质和镉杂质的闪锌矿的电荷差分密度图,它们是通过空位或者杂质原子沿成键方向切出来的。
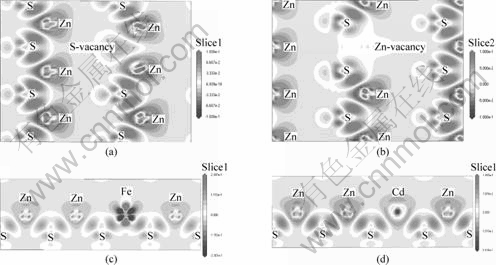
图5 含有硫空位、锌空位、铁杂质和镉杂质的闪锌矿的电荷差分密度图
Fig.5 Plots of differences of charge density contour of ZnS with S-vacancy (a), Zn-vacancy (b), Fe-impurity (c) and Cd- impurity (d)
差分(Δρ)通过公式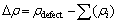 求得。其中:Δρdefect为缺陷体系优化后的总电荷密度分布函数;ρi为体系中优化前的某一个原子的电荷密度分布函数。从图5(a)和(b)可以看出:空位周围的电荷分布发生了明显的变化,锌原子靠近硫空位处失去的电荷显著减少,硫原子靠近锌空位处所得电荷明显减少,这都证明了与硫空位相邻的锌原子的电荷低于其他锌原子的电荷,与锌空位相邻的硫原子的电荷低于其他硫原子的电荷。由图5(c)可见,铁原子在其周围的一个“p轨道形状区域”聚集电荷,而在一个“d轨道形状区域”损失电荷。由图5(d)可见,镉原子与锌原子的电荷分布相似,这主要是因为镉与锌为同族元素,具有相似的电子构型。图6(a)和(b)所示分别为含铁杂质和镉杂质缺陷的闪锌矿的电荷密度图。从图6(a)可以看出:铁硫原子之间的电荷密度明显高于锌硫原子之间的电荷密度,存在较强的电荷密度重叠区。从图6(b)可以看出,镉硫原子之间的电荷密度低于锌硫原子之间的电荷密度,与布居分析结果一致。
求得。其中:Δρdefect为缺陷体系优化后的总电荷密度分布函数;ρi为体系中优化前的某一个原子的电荷密度分布函数。从图5(a)和(b)可以看出:空位周围的电荷分布发生了明显的变化,锌原子靠近硫空位处失去的电荷显著减少,硫原子靠近锌空位处所得电荷明显减少,这都证明了与硫空位相邻的锌原子的电荷低于其他锌原子的电荷,与锌空位相邻的硫原子的电荷低于其他硫原子的电荷。由图5(c)可见,铁原子在其周围的一个“p轨道形状区域”聚集电荷,而在一个“d轨道形状区域”损失电荷。由图5(d)可见,镉原子与锌原子的电荷分布相似,这主要是因为镉与锌为同族元素,具有相似的电子构型。图6(a)和(b)所示分别为含铁杂质和镉杂质缺陷的闪锌矿的电荷密度图。从图6(a)可以看出:铁硫原子之间的电荷密度明显高于锌硫原子之间的电荷密度,存在较强的电荷密度重叠区。从图6(b)可以看出,镉硫原子之间的电荷密度低于锌硫原子之间的电荷密度,与布居分析结果一致。
3 结论
1) 硫空位、锌空位和铁杂质缺陷导致硫化锌晶胞参数变小,镉杂质导致晶胞参数变大。硫空位导致带隙变窄,与硫空位相邻的4个锌原子的电荷明显下降。与硫空位相邻的Zn―S键的布居数减小,共价性减弱,键长变长。锌空位使闪锌矿带隙变宽,使费米能级向低能方向偏移,并在价带上出现简并态,与锌空位相邻的4个硫原子的电荷明显低于其他硫原子的电荷。与锌空位相邻的Zn―S键布居数增大,共价性增强,键长变短。

图6 含有铁杂质和镉杂质的闪锌矿的电荷密度图
Fig.6 Plots of charge density contour of ZnS with Fe-impurity (a) and Cd-impurity (b)
2) 铁杂质使闪锌矿变成n型半导体,使费米能级向高能方向偏移,并在带隙中形成了一个主要由铁的3d轨道贡献的杂质能级。与铁原子相邻的4个硫原子的电荷低于其他硫原子的电荷,Fe―S键比Zn―S键的键长更短,共价性增强。
3) 镉杂质对硫化锌半导体能带结构和态密度影响较小,在硫化锌价带的-7.5 eV处形成一个由镉的4d轨道贡献的能级,Cd―S键布居数下降,共价性减弱,键长变长。
4) 空位缺陷和铁杂质的存在改变了闪锌矿能带结构、电子态密度、电荷分布,从而影响其电化学性质。而镉杂质主要是影响闪锌矿晶格参数和成键性 能,从而更有利于铜的活化。
REFERENCES
[1] 陈建华, 冯其明, 卢毅屏. 电化学调控浮选能带理论及应用(Ⅰ): 半导体-溶液界面能带理论及模型[J]. 中国有色金属学报, 2000, 10(2): 240-242.
CHEN Jian-hua, FENG Qi-ming, LU Yi-ping. Energy band model of electrochemical flotation and its application(Ⅰ): Theory and model of energy band of semiconductor solution interface[J]. The Chinese Journal of Nonferrous Metals, 2000, 10(2): 240-242.
[2] von OERTZEN GU, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. Physics and Chemistry of Minerals, 2005, 32: 255-268.
[3] MARTIN R, BECKER U. First-principles calculations of the thermodynamic mixing properties of arsenic incorporation into pyrite and marcasite[J]. Chem Geol, 2006, 225(3/4): 278-290.
[4] MARC B, MARIA A, JOHN B, KATE W, RICHARD C, CATLOW A. Arsenic incorporation into FeS2 pyrite and its influence on dissolution: A DFT study[J]. Geochimicaet Cosmochimica Acta, 2007, 71(3): 624-630.
[5] 何开华, 余 飞, 姬广富, 颜其礼, 郑澍奎. 第一性原理研究ZnS掺V的光学性质和电子结构[J]. 高压物理学报, 2006, 20(1): 56-59.
HE Kai-hua, YU Fei, JI Guang-fu, YAN Qi-li, ZHENG Shu-kui. Study of optical properties and electronic structure of V in ZnS by first principles[J]. Chinese Journal of High Pressure Physics, 2006, 20(1): 56-59.
[6] 肖 奇, 邱冠周, 胡岳华. 黄铁矿机械化学的计算模拟(Ⅰ): 晶格畸变与化学反应活性的关系[J].中国有色金属学报, 2001, 11(5): 900-904.
XIAO Qi, QIU Guan-zhou, HU Yue-hua. Computational simulation to mechanical activation of pyrite(I): Relation of structural strain to chemistry reaction activity[J]. The Chinese Journal of Nonferrous Metals, 2001, 11(5): 900-904.
[7] 丁宗玲, 郭 英, 邢怀中, 李 伟, 陈效双. PbS电子结构的第一性原理[J]. 东华大学学报, 2008, 34(4): 512-516.
DING Zong-ling, GUO Ying, XING Huai-zhong, LI Wei, CHEN Xiao-shuang. The first principle of the electronic structure of PbS[J]. Journal of Donghua University, 2008, 34(4): 512-516.
[8] LIU H J, CHAN C T. Density functional study on the electronic properties of ZnS: Te[J]. Physics Letters A, 2006, 352: 531-537.
[9] 雷 宇, 胡小强, 刘继东. 硫化锌掺钴对电子结构和光学性质的影响[J]. 南昌大学学报, 2007, 31(6): 564-565.
LEI Yu, HU Xiao-qiang, LIU Ji-dong. Influence of the structural, electronic and optical properties on ZnS doped Co[J]. Journal of Nanchang University, 2007, 31(6): 564-565.
[10] 沈汉鑫, 沈耀文. ZnS掺Mn的电子结构研究[J]. 高压物理学报, 2003, 17(1): 65-68.
SHEN Han-xin, SHEN Yao-wen. Study on electronic structure of ZnS:Mn2+[J]. Chinese Journal of High Pressure Physics, 2003, 17(1): 65-68.
[11] IMAI Y, WATANABE A, SHIMONO I. Comparison of electronic structures of doped ZnS and ZnO calculated by first-principle pseudopotential method[J]. Journal of Materials Science: Materials in Electronics, 2003, 14(3): 149-156.
[12] von OERTZEN GU, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. Journal of Electron Spectroscopy and Related Phenomena, 2005, 144/147: 1244-1247.
[13] TONG Xiong, SONG Shao-xian, HE Jian. Activation of high-iron marmatite in froth flotation by ammoniacal copper(II) solution[J]. Miner Eng, 2007, 20(9): 259-263.
[14] HARMER S L, MIERCZYNSKA V A, BEATTIE D A. The effect of bulk iron concentration and heterogeneities on the copper activation of sphalerite[J]. Miner Eng, 2008, 21(11): 1005-1012.
[15] 熊小勇. 铁成分对硫化锌精矿的半导体性质及化学反应性的影响[J]. 有色金属, 1989, 41(4): 55-66.
XIONG Xiao-yong. Effect of the iron content of zinc sulphide concentrates on their semi-conductivity and chemical reactivity[J]. Nonferrous Metal, 1989, 41(4): 55-66.
[16] SEGALL M D, LINDAN P J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas illustrations and the CASTEP code[J]. J Phys: Cond Matter, 2002, 14: 2717-2743.
[17] PERDEW J P, BURKE K, ERNEZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77: 3865-3868.
[18] VANDERBILT D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism[J]. Phys Rev B, 1990, 41: 7892-7895.
[19] MCMURDIE H F, MORRIS M C, EVANS E H, PARETZKIN B W, WONG N. Methods of producing standard X-ray siffraction powder patterns from the JCPDS research associateship[J]. Powder Diffraction, 1986, 4(1): 334-345.
[20] TRAN T K, PARK W, TONG W, KYI M M, WAGNER B K, SUMMERS C J. Photolumine scence properties of ZnS epilayers[J]. J Appl Phys, 1997, 81: 2803-2809.
[21] ANISIMOV V I, ARYASETIAWAN F, LICHTENSTEIN A I. First-principles calculations of the electronic structure and spectra of strongly correlated systems: The LDA+ U method[J]. J Phys: Cond Matter, 1997, 9: 767-808.
(编辑 何学锋)
基金项目:国家自然科学基金资助项目(50864001)
收稿日期:2009-02-23;修订日期:2009-06-06
通信作者:陈 晔,博士;电话:0771-3233566;E-mail:fby18@126.com

