文章编号: 1004-0609(2006)05-0823-06
Aln(n=2~24, 55)团簇结构特性的第一原理计算
李贵发, 彭 平, 仇治勤, 杨 峰, 韩绍昌
(湖南大学 材料科学与工程学院, 长沙 410082)
摘 要: 采用第一原理赝势平面波方法计算Aln(n=2~24, 55)团簇的几何、 能态与电子结构, 通过结合能Eb、 HOMO-LUMO能隙ΔEH-L与能量二阶差分Δ2E(n)表征和考察团簇原子数n对Aln团簇基态结构稳定性的影响。 结果表明: Aln团簇结构稳定性随团簇原子数n增加而增大, 并在n为7, 11, 13, 19, 23等近幻数和高对称性结构处出现极值, 相对其临近Aln团簇, 具有较高的结构稳定性。 DOS与吸收光谱分析表明, 随着团簇内部s-p电子杂化的逐渐增强, Aln团簇结构的稳定性也随之增加, 但即使当团簇原子数n达到55时, 其微弱的表面效应仍不能消除。
关键词: 赝势平面波方法; Aln团簇; 电子结构; 结构稳定性 中图分类号: O641
文献标识码: A
First-principles calculation for structural character of Aln(n=2-24, 55) clusters
LI Gui-fa, PENG Ping, QIU Zhi-qin, YANG Feng, HAN Shao-chang
(School of Materials Science and Engineering, Hunan University,
Changsha 410082, China)
Abstract: The geometries, energetics and electronic structure of neutral Aln(n=2-24, 55) clusters were calculated using the first-principles pseudo-potential plane wave method. The several parameters, such as binding energy Eb, HOMO-LUMO energy gap ΔEH-L, second difference of energies Δ2E(n) were utilized to characterize and analyze the structure stability of Aln cluster. The results show that the structure stability of Aln clusters increases with the increase of total atom number n. For Al7, Al11, Al13, Al19, Al23 clusters with nearly filled shell of the covalence electron number and high symmetry of geometrical structure, a high structure stability relative to their neighbor Aln clusters can be seen. The analysis of the DOS and absorption spectra shows that the increase of stability of Aln cluster mainly originates from the enhancement of s-p hybridization of Al atoms in cluster, but when up to n=55, the weak influences of surface energy caused by the size of cluster on the properties of Aln cluster can not be eliminated still.
Key words: pseudo-potential plane-wave method; Aln cluster; electronic structure; structural stability
Al团簇由于电子结构简单, 导电性随着原子数目变化而改变, 近年来受到了材料科学工作者的广泛关注[1-6]。 Yang等[1]采用分子动力学(molecular dynamics method, MD) 模拟, 考察了不同原子数Aln(n为2~6, 13, 55, 147) 团簇在温度为500~800K的稳定结构形态, 发现当n为13, 55, 147时, Aln团簇为正二十面体, 并随着原子数的增加, 结合能增大。 最近, 陈莹等[2]通过遗传算法也对Al原子Bernal多面体团簇进行了研究, 发现Al6为正八面体, Al10能量最低结构为两个半八面体覆盖的阿基米德反棱柱。 基于第一原理计算, Rao等[3]采用Gaussian程序计算了Aln(n=2~15)团簇的离化能与结合能, 发现Aln团簇结合能随原子数增加而增大, 但远小于Al晶体的结合能。 Deshpande等[4]采用自旋极化的局域密度近似(local-density approximation, LDA)计算了Aln(n=2~13) 团簇的吸收光谱, 发现当n≤5时, 谱线为分立的离散谱; 从n=6开始谱线成为准连续谱; 但当n=13时, 谱线有一个较强的激发峰。 Li等[5]通过光电谱仪探测了Aln(n=1~162) 团簇的电子结构, 发现从n=7开始, 团簇内部出现s-p杂化, 而这种杂化对小团簇性质的影响起主要作用; 但对n≥75的大团簇, 其影响将逐渐减弱。 Schriver等[6]进一步测定了Aln(n=1~80)的离化能, 发现随着原子数的增加, 团簇离化能逐渐减少, 且在某些特定位置, 如n为7, 14, 17, 23和55等处, 还出现能量谷点, 表明其结构具有相对较高的稳定性。 考虑到上述理论计算与实验研究, 仅仅从不同角度对部分Aln团簇的结构与性质进行研究, 仍缺乏对Aln团簇结构与物性的系统了解, 因此本文作者采用第一原理赝势平面波方法比较全面地计算了Aln(n=1~24, 55)团簇的电子与能态结构, 进而考察了原子数n对Aln团簇结构特性的影响。
1 计算方法与模型
计算程序采用基于密度泛函理论的赝势平面波方法――(Cambridge serial total energy package, CASTEP) 总能计算程序[7]。 采用周期性边界条件, 其晶体波函数由平面波基组展开。 建模采用与Wang等[8]和Pushpa等[9]类似的方法, 即先构建一个晶格常数为2nm的超胞, 然后将团簇模型置于其中, 以此忽略团簇-团簇间的相互影响。 Al团簇初始模型的构建基于Yang等[1]、 Rao等[3]和 Lloyd等[10]的计算结果, 初始Al―Al键长设为dc=0.2863nm, 优化后的能量最低结构模型如图1所示。 参照Rao等[3]的计算与测试结果, 在进行几何优化与总能计算时, 电子交换关联能函数取GGA近似的PBE形式[11], 原子势函数采用Norm-conserving位势[12]。 将Al的3s23p1当作价电子, 其它轨道电子视为芯电子。 在进行各项计算之前, 都用Broyden-Flecher-Goldfarb-Shanno(BFGS)方法进行了几何优化, 以求得它们的局域最稳定结构。 动能截断点取300eV, 布里渊区积分采用Monkhorst-Pack形式的特殊K点方法[13], FFT网格取24×24×24。 自洽计算时, 每个原子的总能量收敛值设为20μeV, 每个原子上的力低于0.5eV/nm, 公差偏移小于0.2pm, 应力偏差小于0.1GPa。

图1 Aln(n=2~24, 55)团簇和Al晶体的计算模型
Fig.1 Calculation models of Aln(n=2-24, 55) clusters and Al crystal
2 结果与讨论
2.1 Al13团簇的测试
选择正二十面体Al13团簇计算了各顶点原子间的键长(d)、 团簇结合能(Eb)与HOMO(Highest occupied molecular orbital)-LUMO(Lowest unoccupied molecular orbital)能隙(ΔEH-L), 结果如表1所列。 从表1可看出, 本研究采用GGA近似计算的d和前人的计算结果符合得很好, 虽然ΔEH-L比Kumar等[14] 采用局域密度近似(LDA)计算结果略大, 但与Yang等[1]采用从头计算(Ab initio)MD模拟得出的结果接近。 由于现在还没有键长和ΔEH-L的实验报道, 但本研究计算的Eb 和实验值与前人的计算结果符合很好。 由于目前还没有关于Al团簇结构的报道, 而采用分子动力学在此方面的研究取得较大的成功, 因此由上述测试可推断本研究所选的计算模型可靠, 且计算条件与方法合适。
表1 Al13团簇顶点原子间键长(d)、 团簇结合能(Eb)与HOMO-LUMO能隙(ΔEH-L)的计算值
Table 1 Calculation value of vertex-to-vertex bond length (d), binding energy (Eb) and HOMO-LUMO energy gap (ΔEH-L) of Al13 cluster

2.2 键长和结合能
为了考察原子数n对Aln团簇几何与能态结构的影响, 首先采用下式计算了Aln(n=2~24, 55) 团簇的平均键长〈R〉和结合能Eb[3]:

式中 Rij为在0.32nm范围内i原子和j原子的距离; nb为在此范围内的原子总数; Etotal(Aln)为Aln团簇的总能量; E(Al)是Al自由原子能量, E(Al)=53.676eV; n表示团簇中的总原子数。 图2(a)所示为平均键长〈R〉随Aln团簇原子数n变化的特性曲线。 图2(b)所示为原子数n对Aln团簇结合能Eb的影响。 由图2(a)可见, 随着原子数n增加, 其键长呈齿形增长, 而且增长速率由快到慢, 并在n为6, 11, 13, 19, 23等外层电子数接近满壳层电子数18, 20, 34, 40, 58, 68处出现峰值, 同时在n=17时也出现了峰值, 但其外层电子数为51, 离满壳层58较远。 由于本文只考虑较高稳定性的幻数结构是否与外层近满壳层的电子有较好的对应, 故对此没有详细的论述。 进一步比较Al55团簇和Al晶体的平均键长发现: 即使是n=55的Aln团簇, 其平均键长〈R〉=0.2753nm也比Al晶体键长dC=0.2863nm小, 表明n=55的Aln团簇在几何结构上也不具有晶体结构特征, 计算结果与Yang等[1]的报道一致。 由图2(b)可见, 本研究计算的结合能Eb比实验值[17]略高。 而Rao等[3] 采用Gaussian程序DFT-GGA近似计算的Eb值则比实验值略低, 但在整体上, 三者变化趋势一致, 即随着原子数n增加, Eb增大, 其结构稳定性也相应随之增大。 值得注意的是, 在n为7, 11, 13, 19, 23等处, 团簇结合能Eb出现了小小的波峰, 由于这几个Aln团簇外层电子数分别为21, 33, 39, 57, 69, 分别与Aln团簇幻数结构的电子数20, 34, 40, 58, 68接近, 表明近幻数结构的Aln团簇相对其它团簇结构结合得更加紧密[3], 而Al55团簇的结合能(Eb=-3.161eV)仍小于Al晶体的结合能(EC=-3.661eV), 说明Aln团簇不如Al晶体结构稳定。
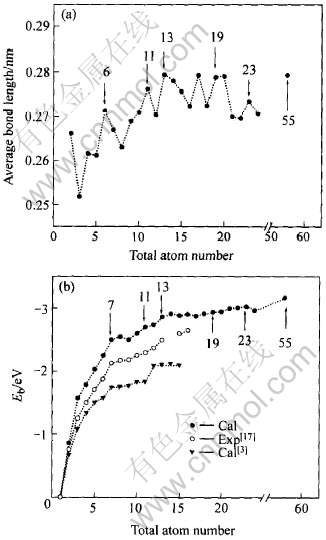
图2 Aln(n=2~24, 55)团簇平均键长〈R〉(a)与
结合能Eb(b)随团簇原子数的变化曲线
Fig.2 Change curves of average band length 〈R〉(a) and binding energy Eb(b) with total atom number n of Aln(n=2-24, 55) cluster
2.3 HOMO-LUMO能隙和能量二阶差分
图3所示为Aln团簇的电子态密度(DOS)曲线。 由图3可见, n≤5的面型小团簇, DOS谱线分立, 当n进一步增加时, DOS谱线逐渐从离散向准连续再到连续谱变化, 表明团簇内部的s-p杂化随n增加而增强[5]。 与Li等[5]的测量结果一致, s-p杂化从n=7开始, 但在n=13处也呈现为分立, 表明Al13团簇具有典型的壳层结构特性, 在其Fermi能级EF以上的0~2eV区域内存在一个较大的HOMO-LUMO能隙ΔEH-L, 与其高电离能的实验结果[6]相对应。 考虑到HOMO-LUMO能隙ΔEH-L随团簇尺寸或原子数的变化对团簇稳定性与电子结构演变(如绝缘体―金属转变)能给出某些有用信息, 如HOMO-LUMO能隙越大, 则预示着相应团簇结构的稳定性越高[18], 因此, 基于图3的DOS曲线, 本文作者画出了Aln团簇的ΔEH-L随团簇总原子数n的变化特性曲线(图4(a))。 由图4(a)可见, 随着n的增加, Aln团簇HOMO-LUMO能隙ΔEH-L总体上趋于减少, 说明其结构稳定性降低, 且金属性增强。 某些特定位如n为7, 13, 18, 23等处出现峰值, 表现出高稳定性, 主要与其近幻数结构特性有关, 在 n=13处出现最高峰, 无疑是由于Al13团簇的几何密堆积结构与外层电子呈近满壳层的结构特性[5]。 但在n为18而非19的近幻数结构处出现峰值, 表明HOMO-LUMO能隙ΔEH-L虽能给出有用信息, 但不是表征结构稳定性的最好参数。

图3 Aln(n=1~24, 55)团簇和Al晶体的态密度曲线
Fig.3 DOS curves of Aln(n=1-24, 55) clusters and Al crystal
由于质谱中的精细结构主要取决于其相邻两团簇结合能之差, 相对稳定的团簇对应着质谱上的峰值位置[18], 因此团簇结构稳定性也可采用团簇总能量相对于原子数n的二阶微商来表示[18]。 为此, 本研究采用下式计算了Aln(n=2~24, 55) 团簇的能量二阶差分Δ2E(n):
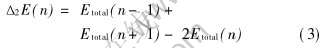
式中 E(n)表示n个原子Aln团簇的总能量。 计算结果如图4(b)所示。 由图4(b)可见, 与上面结合能Eb 的变化趋势(图2(b))一致, 在近幻数结构的n为7, 11, 13, 19, 23等处出现振荡峰, 表明该原子数Aln团簇相对于其它团簇的结构稳定性确实要高一些。 此外, 从图4(b)还不难看出Al14和Al13团簇的Δ2E(n)相当, 说明Al14团簇也具有较高的结构稳定性[15]。 比较这些团簇的结构形态发现, 高稳定性结构的Aln团簇通常对应着一些对称性好的几何形态, 如n=7时构成五角锥形; n=13时为正二十面体, 具有五次轴对称性; n=19时为双二十面体; n=55时为双层闭合二十面体。 可见, 高稳定性的Aln团簇不仅外层电子数接近满壳层电子数, 且具有相对较高的结构对称性。

图4 Aln(n=2~24, 55)团簇HOMO-LUMO能隙ΔEH-L(a)与结合能二阶差分Δ2E(n)(b)随团簇原子数的变化曲线
Fig.4 Change curves of HOMO-LUMO gap ΔEH-L(a) and second difference of
cluster energies Δ2E(n)(b) of Aln(n=2-24, 55) clusters with total atom number
2.4 吸收光谱
图5所示为Aln团簇的吸收光谱。 由图5可看出, 单个Al原子仅有一个电子从s态跃迁到p态产生的尖锐吸收峰, 而Al晶体虽然也只有一个位于约3.5eV处的主峰, 但在高能区和低能区出现了一些由于带间跃迁而引起的吸收增强的精细结构。 与Deshpande等[4]的计算结果一致, 类似于其DOS曲线, 对于n≤6的小团簇, 吸收光谱分立; 当n增加时, 则由准连续到连续谱过渡, 表明吸收光谱也可反映团簇内部的电子结构特性。 此外, 从n=6开始, Aln团簇的吸收光谱在低能区出现了尾巴, 虽然没有半导体团簇[19]那么明显, 但至n=55时, 尾巴仍然存在, 表明体型Aln团簇的表面能即使在n=55时也不能被消除。 因此, Aln团簇的表面效应虽然很弱[17], 但在测量其团簇静态偶极子极化率时却不可忽视表面效应对大团簇的影响。

图5 Aln(n=1~24, 55)团簇和Al晶体的吸收光谱
Fig.5 Absorption spectra of Aln(n=1-24, 55) clusters and Al crystal
3 结论
1) 影响Aln团簇性质的两个主要因素是团簇几何结构和外层电子数。
2) Aln团簇结构稳定性随团簇总原子数n增加而增大, 并在n为7, 11, 13, 19, 23等近幻数和高对称性结构处出现极值, 具有相对较高的结构稳定性。
3) 随原子数n增大, Aln团簇s-p杂化逐渐增强, 而进一步从吸收光谱的分析结果来看, 其微弱的表面效应还仍然存在。
REFERENCES
[1]Yang S Y, Drabold D A, Adams D A, et al. A first-principle local orbital density functional study of Al clusters[J]. Phy Rev B, 1993, 47(3): 1567-1576.
[2]陈莹, 边秀房, 孙民华, 等. 铝原子Bernal 多面体团簇的理论研究[J]. 物理化学学报, 2003, 19(3): 242-245.
CHEN Ying, BIAN Xiu-fang, SUN Min-hua, et al. Theoretical study of bernal polyhedron of aluminum atomic clusters[J]. Acta Phys Chim Sin, 2003, 19(3): 242-245.
[3]Rao B K, Jena P. Evolution of the electronic structure and properties of neutral and charged aluminum cluster: a comprehensive analysis[J]. J Chem Phys, 1999, 111(5): 1890-1904.
[4]Deshpande M D, Kanhere D G. Ab initioan absorption spectra of Aln(n=2-13) cluster[J]. Phys Rev B, 2003, 68(3): 035428-035432.
[5]Li X, Wu H B, Wang X B, et al. s-p hybridization and electron shell structure in aluminum clusters[J]. Phys Rev Lett, 1998, 81(9): 1909-1912.
[6]Schriver K E, Persson J L, Honea E C, et al. Electronic shell of group (Ⅲ)― a metal atomic cluster[J]. Phys Rev Lett, 1990, 64(21): 2539-2543.
[7]Segall M D, Lindan J D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[8]Wang B L, Yin S Y, Wang G H, et al. Structures and electronic properties of ultrathin titanium nanowires[J]. J Phys: Condens Matter, 2001, 13(20): 403-408.
[9]Pushpa R, Narasimhan S, Waghmare U. Symmetries, vibrational instabilities and routes to stable structures of clusters of Al, Sn, and As[J]. J Chem Phys, 2004, 121(11): 5211-5220.
[10]Lloyd L D, Johnston R L. Modelling aluminum clusters with an empirical many-body potential[J]. Chem Phys, 1998, 236(1-3): 107-121.
[11]Calleja M, Rey C, Alemany M M G, et al. Self-consistent density-functional calculations of the geometries, electronic structures, and magnetic moments of Ni-Al clusters[J]. Phys Rev B, 1999, 60(3): 2020-2024.
[12]Hamann D R, Schluter M, Chiang C. Norm-conserving pseudopotentials[J]. Phys Rev Lett, 1979, 43(20): 1494-1497.
[13]Pack J D, Monkhorst H J. Special points for Brillouin-zone integrations―a reply[J]. Phys Rev B, 1977, 16(4): 1748-1749.
[14]Kumar V, Bhattacharjee S, Kawazoe Y. Silicon-doped icosahedral, cuboctahedral, and decahedral clusters of aluminum[J]. Phys Rev B, 2000, 61(12): 8541-8547.
[15]Kumar V. Structure and electronic properties of Al14 and Al13Na clusters[J]. Phys Rev B, 1998, 57(15): 8827-8829.
[16]Gong X G, Kumar V. Enhanced stability of magic clusters: a case study of icosahedral Al12X, X=B, Al, Ga, C, Si, Ge, Ti, As[J]. Phys Rev Lett, 1993, 70(14): 2078-2081.
[17]Ray U, Jarrold M F, Bower J E, et al. Photodissociation kinetics of aluminum cluster ions: determination of cluster dissociation energies[J]. J Chem Phys, 1989, 91(5): 2912-2921.
[18]Wang J L, Wang G H, Zhao J J. Density-functional study of Aun(n=2-20) clusters: lowest-energy structures and electronic properties[J]. Phys Rev B, 2002, 66(3): 035418- 035423.
[19]Vasiliev I, Ogut S, Chelikowsky J R. Ab initio absorption spectra of gallium arsenide clusters[J]. Phys Rev B, 1999, 60(12): 8477-8480.
基金项目: 国家教育部科技重点资助项目(104139); 湖南省自然科学基金资助项目(03-Y3069)
收稿日期: 2005-09-05; 修订日期: 2005-11-21
通讯作者: 彭 平, 教授, 博士; 电话: 0731-8821610; E-mail: ppeng@hnu.cn
(编辑李艳红)

