J. Cent. South Univ. Technol. (2011) 18: 1395-1401
DOI: 10.1007/s11771-011-0852-x
Stability, detonation properties and pyrolysis mechanisms of polynitrotriprismanes C6H6-n(NO2)n (n=1-6)
TANG Zhong-hai(唐忠海)1, 2, OUYANG Yong-zhong(欧阳永中)1,
LIANG Yi-zeng(梁逸曾)1, RAO Li-qun(饶力群)2
1. School of Chemistry and Chemical Engineering, Research Center of Modernization of Chinese Medicines,
Central South University, Changsha 410083, China;
2. College of Bioscience and Biotechnology, Hunan Agriculture University, Changsha 410128, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2011
Abstract: To further test whether polynitriprismanes are capable of being potential high energy density materials (HEDMs), extensive theoretical calculations were carried out to investigate on a series of polynitrotriprismanes (PNNPs): C6H6-n(NO2)n (n=1-6). Heats of formation (HOFs), strain energies (SE), and disproportionation energy (DE) were obtained using B3LYP/6-311+G(2df, 2p)//B3LYP/6-31G* method by designing different isodesmic reactions, respectively. Detonation properties of PNNPs were obtained by the well-known KAMLET-JACOBS equations, using the predicted densities (r) obtained by Monte Carlo method and HOFs. It is found that they increase as the number of nitro groups n varies from 1 to 6, and PNNPs with n?4 have excellent detonation properties. The relative stability and the pyrolysis mechanism of PNNPs were evaluated by the calculated bond dissociation energy (BDE). The comparison of BDE suggests that rupturing the C―C bond is the trigger for thermolysis of PNNPs. The computed BDE for cleavage of C―C bond (88.5 kJ/mol) further demonstrates that only the hexa-nitrotriprismane can be considered to be the target of HEDMs.
Key words: high energy density materials; polynitrotriprismanes; heats of formation; strain energies; disproportionation energy; bond dissociation energy
1 Introduction
In recent years, there has been considerable interest for synthesis and characterization of highly strained molecules [1-4]. Especially for those compact organic cage compounds, they are usually investigated as promising candidates for high energy density materials (HEDMs) when attached to optimum numbers of nitro groups [3, 5-8], due to their superior energetic performances over conventional energetic materials.
A few attempts [5, 8] have been made to demonstrate that triprismane (C6H6) (see Fig.1) can be regarded as a possible candidate for HEDMs when all the hydrogen atoms have been replaced by nitro groups, just from the calculated thermochemical properties of the polynitrotriprismanes (PNNPs) series C6H6-n(NO2)n (n=1-6). However, to be a candidate as HEDMs, not only should the detonation properties meet the criteria of HEDMs (the density (r?1.9 g/cm3), detonation velocities (D?9.0 km/s), and detonation pressure (P?40 GPa)), but also the pyrolysis mechanism and thermal stability should be taken into account [7]. To the best of our knowledge, none of these studies have focused on the pyrolysis mechanism and explosive properties of the polynitrotriprismanes, except for the predicted density and strain energy of the hexa-nitrotriprismane from empirical group additivity method by GILBERT and his co-workers [5].
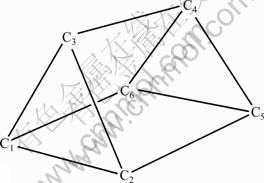
Fig.1 Molecular structure and atomic numbering of triprismane (C6H6)
The aim of this work is to further test whether hexa-nitrotriprismane is capable of being potential HEDMs and which kind of other PNNPs has such properties, through systematical evaluating the heats of formation (HOFs), strain energies (SE), disproportiona- tion energies (DE), density (r), detonation performances and pyrolysis mechanism of PNNPs.
HOF is a key thermodynamic property in chemistry [3, 7], especially important for investigation of the explosive performances of energetic materials. Because it is impractical and dangerous for investigating the HOFs of energetic materials and unstable compounds, computational derived accurate value of HOFs is required. The density functional theory (DFT) B3LYP method using suitable basis sets, can not only produce reliable geometries and accurate energies, but also save more computer resources than those from high level theoretical methods. Therefore, the HOFs of all PNNPs were calculated at B3LYP/6-311+G (2df, 2p)//B3LYP/ 6-31G* level of DFT theory by designing isodesmic reaction.
Strain energy is the difference between the observed heat of formation and that expected from a strain free molecule with the same number of atoms [9]. It has proven to be very valuable quantity to examine the balance of stabilization and destabilization effects that manifest themselves in the “measured” strain energy of cyclic molecules [10]. The SE of PNNPs was obtained using the reference of triprismane as a criterion by means of isodesmic reaction.
Repulsion interactions among nitro groups were predicted by the calculated disproportionation energies (DE). In addition, D and P were obtained using empirical KAMLET-JACOBS equation [11] with the density functional theory based density (r) and HOFs, which are essential for evaluating the detonation properties of energetic compounds. Bond dissociation energies for C― C and C―N bond were calculated and compared to investigate the pyrolysis mechanism and thermal stability.
2 Computational methods
All calculations were carried out with the Gaussian 03 system of programs [12]. The full optimized geometries and zero-point energy corrections were calculated at Bech3-Lee-Yang-Parr (B3LYP) hybrid density functional level of theory, with 6-31G* basis. Single-point energy calculations on the optimized geometries were performed using the relatively higher level B3LYP/6-311+G (2df, 2p). The thermal corrected values and the zero-point energies (ZPE) were scaled by 0.98.
The isodesmic reaction approach [13], in which the HOFs can be estimated by simply calculating the reaction energy or reaction enthalpy with quantum chemical methods, has been employed very successfully to derive the HOFs [14]. The heats of formation of all PNNPs at 298 K, ?H298, were evaluated by the following expressions:
C6H6-n(NO2)n+nCH4?C6H6+nCH3NO2 (1)

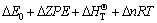 (2)
(2)
where  and
and 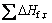 are the sums of the heats
are the sums of the heats
of formation of the products and the reactants,
respectively.  is the enthalpy change of the reaction
is the enthalpy change of the reaction
between the products and reactants at 298 K. ? E0, ?ZPE,
and  are the changes of the total energy, the zero-
are the changes of the total energy, the zero-
point energies, and the thermal correction on going from 0 to 298 K between the products and reactants, respectively. It should be noted that ?PV equals ?nRT for the reaction in gas phase, and for the isodesmic reaction (1), ?n=0, so ?PV=0.
The SE can be regarded as the relative SE of PNNPs plus the known SE value of triprismane. Because of the lack of experimental value, the SE of the reference compound for triprismane (606.9 kJ/mol) was derived from Ref.[15], which was calculated by G3/B3LYP method based on the isodesmic reaction. In addition, the relative SE values of PNNPs were calculated via the known SE values of triprismane as a criterion [15], i.e., the available SE of triprismane was taken as a reference compound, and the ring strain energy was computed according to the total energy changes of the following isodesmic reactions, with correction of zero-point vibrational energy:
C6H6+n(CH3)3C?n(CH3)3CH+C6H6-nXn (3)
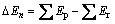 (4)
(4)
where n is the number of nitro groups in PNNPs. From the isodesmic reaction (3), the relative SE of PNNPs can be calculated using Eq.(4).
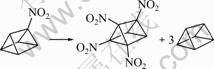
Isodesmic disproportionation reactions [16] are designed to quantitatively investigate the strength of the interactions among the nitro groups. N-molecules of mono-nitrotriprismane can be disproportioned to triprismane (NO2)n plus (n-1) molecules of triprismane. For example, the 1,2,3,4-trinitrotriprismane can be obtained as
 (5)
(5)
The disproportionation energy is defined as total energy change in Eq.(6), corrected by zero-point energy between products and reactants:
?E=?E0+?ZPE (6)
where ?E0 and ?ZPE are the changes in total energy and zero-point energy of the products and reactants at 0 K.
The empirical KAMLET-JACOBS equations were used to estimate the values of D and P for the explosive compounds containing C, H, O, and N as follows:

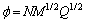 (7)
(7)
 (8)
(8)
where P, D, N, M and r0 denote the detonation pressure (GPa), the detonation velocity (km/s), the amount of substance of detonation gases per gram of explosive, the average relative molecular mass of these gases, and the density of explosive (g/cm3), respectively; Q, the chemical energy of detonation (kJ/mol), is defined as the difference of the HOFs between the products and reactants, based on the most exothermic principle [12]. The theoretical density of each PNNP was obtained from the relative molecular mass divided by the average molecular volume (V). The average molar volume of each compound was obtained from the statistical molar volume of 100 molar volumes [17]. The molar volume of each molecule, defined as the volume inside a contour of 0.001e/Bohr3 density, was computed by Monte Carlo method in the Gaussian 03 program package. It has been demonstrated that the theoretically derived density is in good agreement with experimental density and describes the explosive phenomena well [7].
The hemolytic bond dissociation energy [18] at 0 K can be obtained in terms of Eq.(9):
D0(AB)=E0(A)+E0(B)-E0(AB) (9)
where E0=Eelec+ZPE (Eelec and ZPE denote the electronic energy and zero-point energy, respectively).
3 Results and discussion
3.1 Heats of formation
To calculate the HOFs for PNNPs, the HOF of a reference compound for parent triprismane C6H6 should be known in the isodesmic reaction (1). However, to the best of our knowledge, there is no available experimental HOF. In this work, the isodesmic reaction Eq.(10), in conjunction with the calculated enthalpy HT values from the high-level ab initio G3 method for the reference compound (listed in the last column of Table 1), is designed to calculate its HOF:
C6H6+12CH4?9C2H6 (10)
The calculated HOF of parent triprismane, with a large value of 560.7 kJ/mol, can be comparable to the previously reported results (559.8 kJ/mol [19] and 565.2 kJ/mol [16]). This demonstrates the reliability of the method used for the calculation of HOF by combination of the isodesmic reaction and G3 theory.
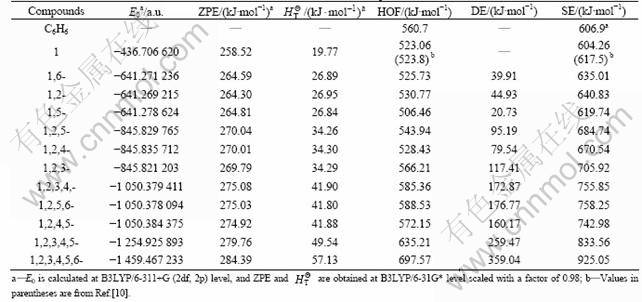
Table 1 lists the theoretical total energies (E0), zero- point energies (ZPE), thermal corrections and experimental HOFs for the reference compounds. Based on the energy properties of the reference compounds given in Table 1, the HOFs of PNNPs can be obtained by combination with DFT method and isodesmic reaction, as listed in Table 2. It is surprising from Table 2 that the calculated HOF of mono-nitrotriprismane (523.06 kJ/mol) is in good agreement with the available theoretical value (523.8 kJ/mol) derived from G3/B3LYP method [15]. This implies that the performance of the DFT theory with isodesmic reaction is as good as the computationally demanding G3 theory in the calculation of HOFs. For the first six PNNPs, the HOFs is smaller than that of triprismane. This is mainly due to the strong electron- withdrawing effect of the nitro groups, which stabilizes the skeleton cage of triprismane. However, from 1,2,3-tritriprismane to hexa-nitrotriprismane, the HOFs increase dramatically with the number of nitro groups increasing, which is attributed to the strong interactions among more nitro groups over electron-withdrawing effect. The HOF of hexa-nitrotriprismane with 697.57 kJ/mol is even as large as that of octa-nitrocubane (726.47 kJ/mol) [3], implying that the introduction of nitro groups is the main energy origin of the PNNPs series and the PNNPs are capable of having better energy properties as more energetic compound of octa- nitrocubane, at least better than other energetic compounds such as TNT, and HMX.
and experimental HOFs for the reference compounds. Based on the energy properties of the reference compounds given in Table 1, the HOFs of PNNPs can be obtained by combination with DFT method and isodesmic reaction, as listed in Table 2. It is surprising from Table 2 that the calculated HOF of mono-nitrotriprismane (523.06 kJ/mol) is in good agreement with the available theoretical value (523.8 kJ/mol) derived from G3/B3LYP method [15]. This implies that the performance of the DFT theory with isodesmic reaction is as good as the computationally demanding G3 theory in the calculation of HOFs. For the first six PNNPs, the HOFs is smaller than that of triprismane. This is mainly due to the strong electron- withdrawing effect of the nitro groups, which stabilizes the skeleton cage of triprismane. However, from 1,2,3-tritriprismane to hexa-nitrotriprismane, the HOFs increase dramatically with the number of nitro groups increasing, which is attributed to the strong interactions among more nitro groups over electron-withdrawing effect. The HOF of hexa-nitrotriprismane with 697.57 kJ/mol is even as large as that of octa-nitrocubane (726.47 kJ/mol) [3], implying that the introduction of nitro groups is the main energy origin of the PNNPs series and the PNNPs are capable of having better energy properties as more energetic compound of octa- nitrocubane, at least better than other energetic compounds such as TNT, and HMX.
The relative distance of nitro groups also has some effects on the HOFs of PNNPs, and thus influences the stability of the compounds, especially apparent for the isomers with the same number of substitutions. The shorter the distance between nitro groups is, the stronger the repulsive energy (DE) is (see Table 2), the larger the corresponding HOF of the compound is, and the less stable the PNNPs will be. For example, as for the isomers for the isomers with two nitro groups, the HOF of 1,2-dinitrotriprismane is larger than that of 1,6- dinitrotriprismane and 1,5-dinitrotriprismane, due to the relatively shorter distance between nitro groups in 1,2- dinitrotriprismane compared to that of other two isomers. Therefore, the relative HOFs order of PNNPs can be distinguished, based on the number and relative position of the substitutions, which is very useful for estimating the relative thermal stability of PNNPs for isomers.
Table 1 Theoretical total energies (E0), zero-point energies (ZPE), thermal corrections  experimental HOFs, and enthalpy values from G3 theory for reference compoundsa
experimental HOFs, and enthalpy values from G3 theory for reference compoundsa

Table 2 Total energies (E0), zero-point energies (ZPE), thermal correctionsHOF, SE and DE for title compoundsa
3.2 Strain energy
Actually, the strain energy is also a very important parameter for the choice of the HEDMs, due to the fact that it is directly related to the stability and energetic performance of the title compound.
Table 2 also shows the SE of PNNPs predicted at B3LYP/6-311+G (2df, 2p)//B3LYP/6-31G* level by designing isodesmic reaction (3). The SE value of mono- substituted nitro compound (604.26 kJ/mol) can be comparable to that derived from G3/B3LYP method (617.5 kJ/mol) [15]. The difference between them is only about 13 kJ/mol, which can be attributed to the higher accuracy of the G3/B3LYP method compared to the DFT theory in calculation of total energies. However, the G3/B3LYP calculations are computationally demanded for large systems when more nitro-substituents are attached to the triprismane. The SE values increase very slightly at the beginning, but then increase rapidly as the number of the substituted groups increases. This further demonstrates that NO2 groups are more electronegative than the withdraw electrons from C atom of triprismane, which firstly reduce the repulsion between C―C bonds and result in the release of the strain of the skeleton. With more numbers of H atoms substituted by NO2 groups, the repulsion interactions become predominant and repulsion energies increase sharply, leading to large strains of the skeleton. The SE of hexa-nitrotriprismane (925.05 kJ/mol) can be comparable to that of octa- nitrocubane (1 075.09 kJ/mol) [20], which indicates that the hexa-nitrotriprismane is capable of being the candidates of HEDMs.
3.3 Detonation properties
Table 3 presents the calculated average molecular volume (Vm), density (r), chemical energy of detonation (Q), detonation velocities (vd), and detonation pressure (P) of the PNNPs. It has been demonstrated [7] that the derived density in the present study has been successful for evaluating theoretical densities for such molecular systems. Furthermore, the calculated density (r= 2.147 g/cm) and detonation pressure (P=52.939 3 GPa) for hexa-nitrotriprismane in this work can be compared to the previously reported values [6] r=2.138 g/cm3 and P=49.3 GPa, respectively, obtained by empirical group additively method, in which the relative errors for calculated density is only 0.4%. This further confirms the applicability of the present method, which can be applied to evaluate the explosive properties for these molecular systems. However, there may be some deviations from the experimental data, to some extent, but they are the most reliable theoretical method available currently for estimating the explosive properties, which are very important properties for characterization on energetic materials.
From Table 3, it can be seen that with the number of nitro groups increasing from n=1 to 6, Vm, r, Q, vd, and P of all corresponding PNNPs increase. Moreover, Vm, r, Q, vd, and P are in good linear relationship with the number of the nitro substitutions. Figures 2(a)-(e) display the corresponding correlations with coefficients of 0.996 8, 0.981 5, 0.977 0, 0.982 4 and 0.994 9, respectively. It is worth noting that, for the isomers with the same number of substituted groups, the most stable compound with the least detonation values were chosen for analysis. The density of all PNNPs (shown in Table 3) has larger values (r=1.623 7-2.147 1 g/cm3) compared to that of the parent triprismane (r=1.255 4 g/cm3), except for the mono-substitution compound. Particularly, PNNPs with n?4 have excellent detonation performances (r? 9.146 9 g/cm3, Q? 2 029.92 J/g, vd?9.146 9 km/s, P? 38.457 1 GPa), which meet the standard as HEDMs, and most likely become the candidates for HEDMs.
Table 3 Average molecular volume, densities, detonation velocities, and pressures for polynitroprismanes
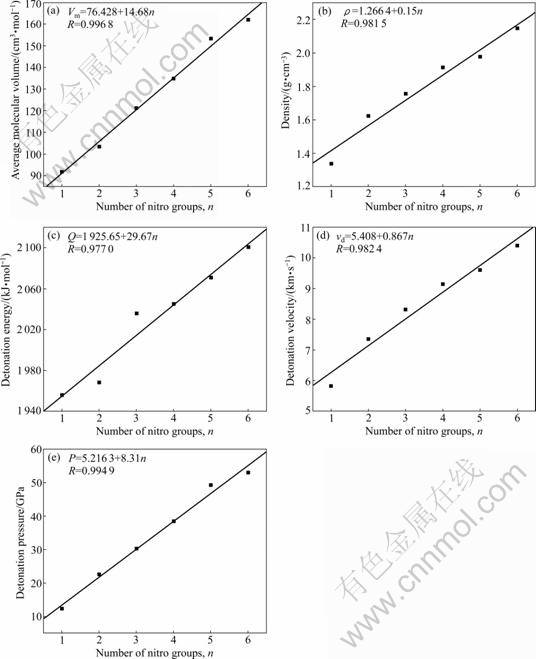
Fig.2 Linear relationship between number of nitro groups and average molecular volume (a), densities (b), detonation energy (c), detonation velocity (d) and detonation pressures (e), respectively, for PNNPs
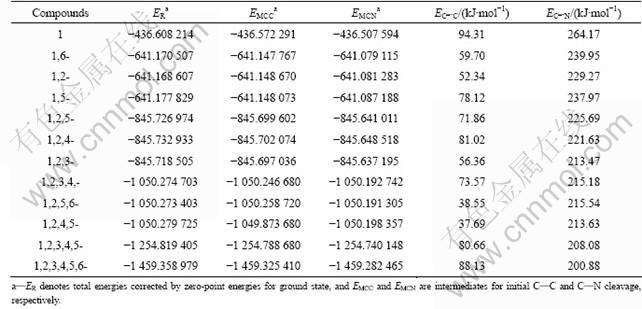
Table 4 Total energies and bond dissociation energies (BDE) for C―C (EC―C) and C―N (EC―N) bonds at 0 Ka
3.4 Pyrolysis mechanism and thermal stability
Besides the discussion above, the bond dissociation energies (BDE) for each possible trigger bond are used to investigate the pyrolysis mechanism and thermal stability for PNNPs. Generally speaking, the smaller the BDE for breaking a bond is, the more easily the bond is broken. In this work, the BDE for two possible initial steps in the pyrolysis route are calculated (Table 4): 1) breaking a C―NO2 bond and 2) breaking a C―C bond on the cage skeleton. It should be noted that the weakest C―C or C―NO2 bonds based on the Mulliken population analyses are chosen as the initially breaking bonds.
It is found, from Table 4, that the BDE for dissociation of C―NO2 bond for each PNNP bond is much larger than that for breaking C―C cage skeleton, indicating that the rupture of C―C bonds should be the trigger bond during the thermolysis initiation process. This is the same as the pyrolysis process of octa-nitrocubane, but it is different from that of other energetic compounds such as TNT, RDX, and HMX, in which initial dissociation reaction occurs between the ring and NO2.
It has been recommended by CHUNG et al [21] that to be candidates for potential HEDMs, the energy barriers to decomposition and bimolecular destruction should be more than 20 kcal/mol (84 kJ/mol). It can be seen from Table 4 that the hexa-nitrotriprismane meets the needs, with the BDE of 21.08 kcal/mol (88.54 kJ/mol). Therefore, from the analysis mentioned above, only the hexa-nitrotriprismane of all PNNPs satisfies the standard as candidates for HEDMs, although other PNNPs with more than four nitro groups have better detonation properties.
4 Conclusions
1) The calculated HOFs, DE and SE suggest that the introduction of NO2 groups to the triprismane is indeed the source of the energy. They are correlated with each other, which reflect the relative stability of PNNPs, especially useful for identifying the isomers with same nitro groups.
2) The detonation properties of PNNPs increase with the number of nitro groups increasing from n=1 to 6, and moreover, they are in good linear relationship with the number of the nitro substitutions. However, only when n?4 can PNNPs be regarded as energetic compounds.
3) The comparison of BDE shows that homolysis of the C―C bond is predicted to be the trigger bond during thermolysis.
4) The BDE obtained from B3LYP/6-311+G (2df, 2p)//B3LYP/6-31G* calculations, in conjunction with the detonation properties, further confirms that only the hexa-nitrotriprismane can be considered to be the target of HEDMs.
References
[1] Harris N J, Lammertsma K. Tautomerism, ionization, and bond dissociations of 5-Nitro-2,4-Dihydro-3H-1,2,4-Triazolone [J]. Journal of the American Chemical Society, 1996, 118(34): 8048-8055.
[2] Kortus J, Pederson M R, Richardson S L. Density functional-based prediction of the electronic, structural, and vibrational properties of the energetic molecule: Octanitrocubane [J]. Chemical Physics Letters, 2000, 322(3): 224-230.
[3] Zhang Ji, Xiao He-ming. Computational studies on the infrared vibrational spectra, thermodynamic properties, detonation properties, and pyrolysis mechanism of octanitrocubane [J]. Journal of Chemical Physics, 2002, 116(24): 10674-10683.
[4] Bach R D, Dmitrenko O. Strain energy of small ring hydrocarbons. Influence of C―H bond dissociation energies [J]. Journal of the American Chemical Society, 2004, 126(13): 4444-4452.
[5] GIBERT P S, JACK A, EVERETT E G,OSCAR S,NORMAN S. Research towards novel energetic materials [J]. Journal of Energetic Materials, 1986, 4(1/2/3/4): 5-28.
[6] Eaton P E, Gilardi R L, Zhang M X. Polynitrocubanes: Advanced high-density, high-energy materials [J]. Advanced Materials, 2000, 12(15): 1143-1148.
[7] XU X J, XIAO H M, GONG X D, ZHU W H. Computational studies on polynitrohexaazaadmantanes as potential high energy density materials [J]. The Journal of Physical Chemistry A, 2006, 110(17): 5929-5933.
[8] KAUSTUBh A J, SHRIDHAR P G. Molecular electrostatic potentials and electron densities in nitrotriprismanes [J]. Journal of Molecular Structure: THEOCHEM, 2005, 724(1/2/3): 87-93.
[9] Kenneth B W. The Concept of strain in organic chemistry [J]. Angewandte Chemie International Edition in English, 1986, 25(4): 312-322.
[10] Kitchen D B, Jackson J E, Allen L C. Organosilicon rings: Structures and strain energies [J]. The Journal of the American Chemical Society, 1990, 112(9): 3408-3414.
[11] KAMLET M J, JACOBS S J. Chemistry of detonations. I. A simple method for calculating detonation properties of C-H-N-O explosives [J]. Journal of Chemical Physics, 1968, 48(1): 23-35.
[12] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian IncWallingford CT [Z]. 2004.
[13] GEORGE P, TRACHTMAN M, BOCK C W, BRETT A M. Empirical resonance energies for benzene and pyridine [J]. Tetrahedron Letters, 1985, 26(46): 5667-5670.
[14] WIBERG K B, OCHTERSKI J W. Comparison of different ab initio theoretical models for calculating isodesmic reaction energies for small ring and related compounds [J]. Journal of Computational Chemistry, 1997, 18(1): 108-114.
[15] Novak I. Substituent effects on steric strain [J]. Chemical Physics Letters, 2003, 380(9): 258-262.
[16] Hrovat D A, Borden W T, Eaton P E, Kahr B. A computational study of the interactions among the nitro groups in octanitrocubane [J]. Journal of the American Chemical Society, 2001, 123(7): 1289-1293.
[17] Wong M W, Wiberg K B, Frisch M J. Ab initio calculation of molar volumes: Comparison with experiment and use in solvation models [J]. Journal of Computational Chemistry, 1995, 16(3): 385-394.
[18] Blanksby S J, Ellison G B. Bond dissociation energies of organic molecules [J]. Accounts of Chemical Research, 2003, 36(4): 255-263.
[19] CHEUNG T S, LAW C K, LI W K. Gaussian-3 heats of formation for (CH)6 isomers [J]. Journal of Molecular Structure: THEOCHEM, 2001, 572(1/2/3): 243-247.
[20] FAN Xiao-wei, JU Hai-xue, XIA Qi-ying, XIAO He-ming. Strain energies of cubane derivatives with different subsituent groups [J]. Journal of Hazardous Materials, 2008, 151(1): 255-260.
[21] CHUNG G S, SCHMIDT M W, GORDON M S. An Ab initio study of potential energy surfaces for N8 isomers [J]. The Journal of Physical Chemistry A, 2000, 104(23): 5647-5650.
(Edited by HE Yun-bin)
Foundation item: Projects(2006DFA41090, 2007DFA40680) supported by the International Cooperation Project on Traditional Chinese Medicines of Ministry of Science and Technology of China; Project(20475066) supported by the National Natural Science Foundation of China
Received date: 2010-05-18; Accepted date: 2010-12-30
Corresponding author: LIANG Yi-zeng, Professor; Tel:+86-731-88830824; E-mail: yizeng_liang@263.net

