网络首发时间: 2018-03-13 13:01
稀有金属 2018,42(11),1193-1198 DOI:10.13373/j.cnki.cjrm.xy17120004
NiF2 掺杂对MgH2 解氢性能影响机制研究
刘友成 周兵 刘绍忠
邵阳职业技术学院汽车与智能制造学院
湖南大学汽车车身先进设计制造国家重点实验室
摘 要:
采用第一原理赝势平面波方法, 计算分析了NiF2 掺杂对MgH2 解氢性能的影响, 合金形成热计算结果表明:F原子优先占据MgH2 中的H原子位, 无论F原子占H位还是占居间隙位置都能形成更为稳定的结构。F原子占据MgH2 间隙位置时, H原子的解离能明显降低, 说明F原子占据间隙位置时, 能够改善MgH2 体系的解氢性能。Ni原子置换MgH2 中的Mg原子后, MgH2 体系的形成热与H原子的解离能都降低, 说明在MgH2 体系中加入Ni原子能够改善MgH2 体系的解氢性能。电子态密度, 交叠聚居数结果表明:F或Ni原子掺杂后MgH2 解氢能力增加, 归因于MgH2 体系费米能级附近的能隙变窄以及H-H原子的键长变短。合金形成热及交叠聚居数结果表明, Mg与F元素容易成键形成更为稳定的Mg F2化合物, Ni元素与MgH2 形成不稳定的Mg2Ni H4化合物, 从微观层面证实了NiF2 的加入促进化学反应NiF2 +3MgH2=Mg F2+Mg2Ni H4+H2的进行, 形成更为稳定的Mg F2和不稳定的Mg2Ni H4化合物, 从而改善MgH2 体系的解氢性能。
关键词:
NiF2掺杂 ;MgH2 ;解氢性能 ;赝势平面波 ;
中图分类号: TB34;TQ116.2
作者简介: 刘友成 (1976-) , 男, 湖南邵阳人, 硕士, 副教授, 研究方向:材料性质的理论模拟;电话:13036734206;E-mail:liuyoc@163.com;
收稿日期: 2017-12-06
基金: 国家自然科学基金项目 (51275162); 湖南省教育厅科研项目 (16C1477); 2017年国内访问学者项目资助;
Affecting Mechanism of NiF2 Doping on Improvement of Dehydrogenating Properties of MgH2 Systems
Liu Youcheng Zhou Bing Liu shaozhong
Automobile and Intelligent Manufacture College, Shaoyang Vocational and Technical College
State Key Laboratory of Advanced Design and Manufacture for Vehicle Body, Hunan University
Abstract:
A first-principles plane-wave pseudopotential method was used. The mechanism of NiF2 doping on the improvement of dehydrogenating properties of MgH2 systems was studied. Alloy formation heat calculation results showed that F atom preferred to occupy the H atom site of MgH2 , whether F atom replaced H atom or occupied interstitial site MgH2 could form a more stable structure. When F atom occupied the interstitial site of MgH2 , the dissociation energy of H atom was obviously reduced. It meant that when the F atom occupied the interstitial site, the dehydrogenation properties of the MgH2 system were improved. When Ni atom replaced Mg atom, both the formation heat of the MgH2 system and the dissociation energy of H atom were reduced. It meant that after Ni atom replaced Mg atom, the dehydrogenation properties of the MgH2 system were improved. After analyzing the densities of states ( DOS) and overlap populations, it was found that after doped with F or Ni atom the dehydrogenation properties of MgH2 were increased, it was attributed to the narrow of the energy gap near the Fermi energy level and the shortening of H-H bond length. The calculation results of alloy formation heat and overlap populations showed that Mg and F elements tended to form more stable MgF2 compound, and Ni elements and MgH2 formed unstable Mg2 Ni H4 compound. It showed that the addition of NiF2 could accelerate the chemical reaction of NiF2 + 3 MgH2 =MgF2+ Mg2 Ni H4+ H2, a more stable MgF2 and unstable Mg2 Ni H4 compound were formed to improve the hydrogenation of the MgH2 system.
Keyword:
NiF2 doping; MgH2; dehydrogenation property; plane-wave pseudopotential method;
Received: 2017-12-06
镁基储氢材料的储氢量高、成本低、质量轻, 受到研究人员的广泛关注。其中MgH2 的理论储氢量高达7.6% (质量分数) , 是一种很有诱惑力的金属储氢材料, 但MgH2 在热力学上过于稳定, 而动力学过于缓慢限制了它的实际应用
[1 ]
。研究人员通常在MgH2 体系内加入少量金属及其化合物作为催化剂来改善MgH2 的吸放氢性能
[2 ,3 ]
。Zaluski等
[4 ]
采用机械合金化的方法在MgH2 中加入镍制备MgNi2 合金, 加快了MgH2 的氢化反应速度。Suda
[5 ]
通过对Mg及其合金进行氟化处理, 发现氟化处理后Mg合金表现出良好的吸氢性能。周惦武等
[6 ]
采用第一原理方法, 分析了Ni元素改善MgH2 体系解氢能力的理论机制。袁江等
[7 ]
采用第一原理计算分析, 发现Ti F3 能够改善MgH2 的解氢性能。张健等
[8 ]
采用球磨法在MgH2 中复合添加K2 Ti6 O13 与Ni, 改善了MgH2 的解氢性能。Jin等
[9 ]
在MgH2 中加入Ni F2 , FeF2 , Ti F3 等金属氟化物作催化剂, 发现Ni F2 能够提高MgH2 体系解氢性能, 并根据实验中出现的MgF2 , 推测Ni F2 的加入加速了化学反应Ni F2 +3MgH2 =MgF2 +Mg2 Ni H4 +H2 的进行。然而, 关于Ni F2 对MgH2 体系解氢性能的理论机制研究, 尚不多见。本文采用第一原理赝势平面波方法, 探讨Ni F2 掺杂后, F, Ni元素对MgH2 体系解氢性能影响的微观机制, 期望为MgH2 材料的实际应用提供理论指导。
1计算模型与方法
MgH2 晶体的单胞模型如图1 (a) 所示, 晶格常数分别为a=0.4501 nm, c=0.3010 nm。空间群为P42/mnm (No.136) 。MgH2 晶胞由2个Mg原子和4个H原子组成, 2个Mg原子坐标为 (0, 0, 0) ;4个H原子坐标为 (0.304, 0.304, 0)
[7 ]
。计算时选取1×3×1的超胞模型Mg6 H12 图1 (b) 所示, Ni原子占Mg原子位置时形成Mg5 H12 Ni超胞, 图1 (c) 所示, F原子占H原子位置时形成Mg6 H11 F超胞, 图1 (d) 所示, F原子占间隙位置时形成Mg6 H12 F超胞, 图1 (e) 所示, 由于F原子的半径较小, 不考虑F原子占Mg位的情况。
采用Material Studio软件中CASTEP模块进行计算。基于第一原理赝势平面波方法, 交换关联能函数采用GGA中的PBE形式
[10 ]
, 赝势取超软 (ultrasoft) 赝势。计算选取1×3×1的Mg6 H12 超胞。在进行能量计算之前, 采用BFGS方法对超胞进行几何优化
[11 ]
, 以求得它们的局域最稳定结构。超胞总能量收敛值为1.0×10-5 e V・atom-1 (Fine) , 每个原子的力为0.003 e V・nm-1 (Fine) , 公差偏移为1.0×10-4 nm (Fine) , 应力偏差取0.05 GPa (Fine) 。
2结果与讨论
2.1晶格参数
MgH2 晶体优化后的平衡晶格常数a=b=0.4534 nm, c=0.3021 nm, 与周惦武等
[6 ]
的计算值a=0.4534 nm, c=0.3020 nm相吻合, 与实验值a=0.4501 nm, c=0.3010 nm的误差小于0.8%, 气态H2 分子优化后的键长为0.0750 nm, 与实验值
[12 ]
(0.0741 nm) 的误差为1.08%, 与李闯等
[13 ]
的计算结果一致, 气态F2 分子优化后的键长为0.1418nm, 与李闯等
[13 ]
的计算值0.1422 nm, Yin等
[14 ]
的计算值0.1424 nm误差小于0.5%, 计算结果说明计算条件与参数的选用符合要求。
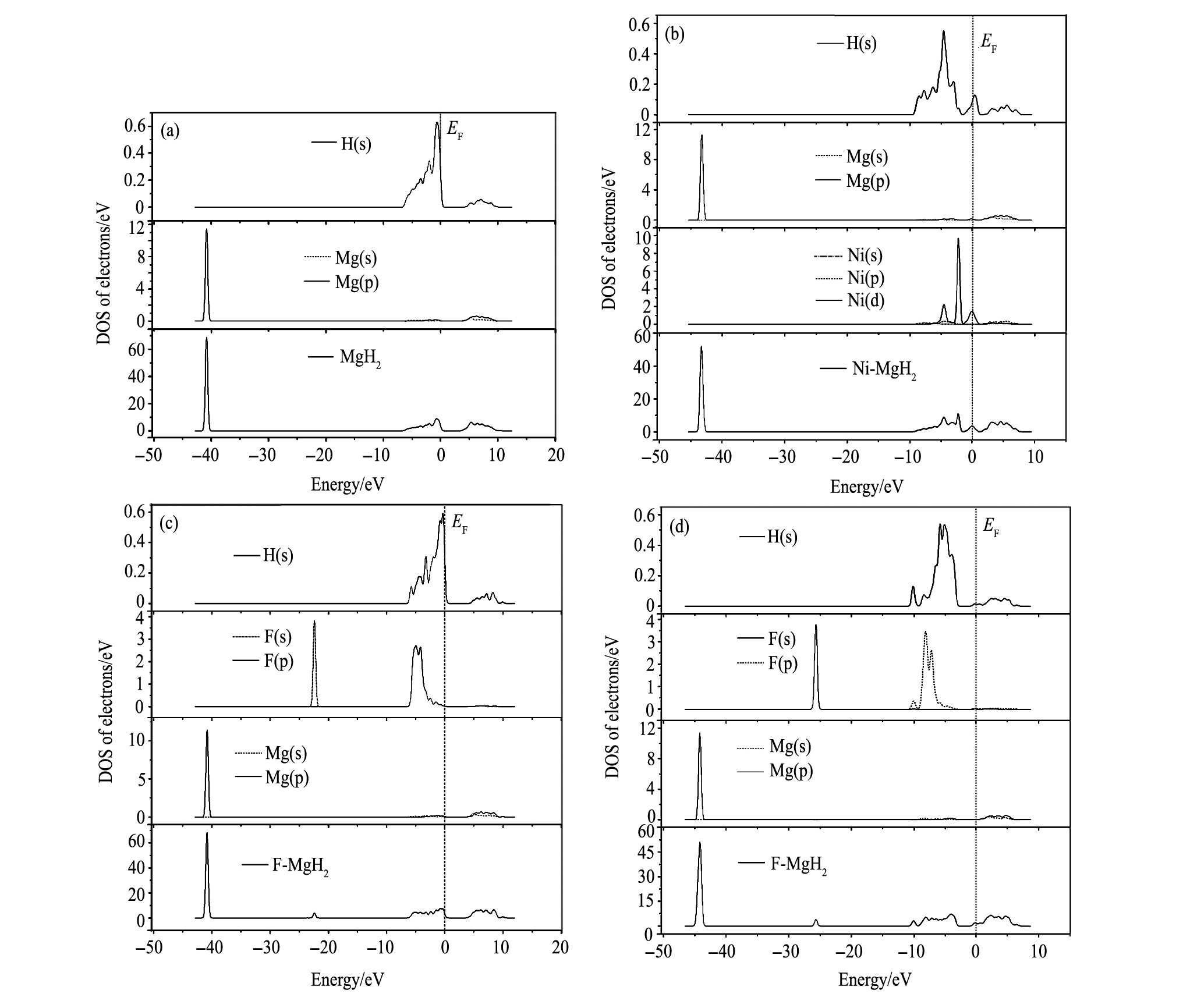
图1 MgH2晶体结构模型及其超胞计算模型Fig.1 MgH2crystal structure model and supercell model
(a) MgH2 ; (b) Mg6 H12 ; (c) Mg5 H12 Ni; (d) Mg6 H11 F; (e) Mg6 H12 F
2.2合金的形成热
储氢合金的吸放氢热力学性能通常用 (P-C-T) 来描述, Van't Hoff关系式如下
[15 ]
式中, po 为标准大气压, p为解氢平台压;ΔH为合金的形成热;ΔS为熵变;T为温度;R为摩尔气体常数。由MgH2 在吸放氢平台内ΔS为定值可得:
由 (2) 式可知, 在温度一定的情况下 (ΔH) 的大小决定着解氢平台压的高低。合金形成热 (ΔH) 越小, 合金体系的结构稳定性越差, 解氢能力就越强。因此, 可以通过计算合金形成热 (ΔH) 来预测金属化合物MgH2 的解氢能力。MgH2 体系每摩尔氢分子的合金形成热 (ΔH) 计算公式如下:
式中, Etot abc 表示超胞的总能量, Es a , Es b 分别表示平衡状态下Mg, H原子的能量, Es c 表示平衡状态下F或Ni原子的能量, m, n, p表示每种原子的原子数量。根据式 (3) 计算的超胞形成热见图2所示, 形成热 (ΔH) 大小顺序为:Mg6 H11 F>Mg6 H12 F>Mg6 H12 >Mg5 H12 Ni。Mg6 H12 形成热的计算值为-64.527k J・mol-1 , 比Zhou等
[16 ]
的计算值-62.301 k J・mol-1 略大, 比实验值
[17 ]
(-76.15±9.2) k J・mol-1 略小。
Mg6 H11 F与Mg6 H12 F的形成热大于Mg3 H6 的形成热, 说明F原子优先占H位, 无论F原子占H位还是占居间隙位置都能形成更为稳定的结构;Mg5 H12 Ni的形成热小于Mg3 H6 的形成热, 说明Ni原子占Mg位, 能够降低MgH2 合金的稳定性。结合J
[9] 2 中加入Ni F2 球磨后出现Mg2 Ni H4 , 可知Ni与MgH2 化合后形成Mg2 Ni H4 , Mg2 Ni H4 形成热的
[6] -1 , 比MgH2 的
[
17] (-76.15±9.2) k J・mol-1 要低, 说明Mg2 Ni H4 的稳定性较差, 有利于解氢的进行。合金形成热计算结果表明, 加入F元素MgH2 体系的稳定性增加, 加入Ni元素MgH2 体系的稳定性降低, 较好的解析了J
[9] 2 推测Ni F2 与MgH2 化合后形成更为稳定的MgF2 和不稳定的Mg2 Ni H4 化合物。
图2 MgH2合金的形成热Fig.2 Formation heat of MgH2alloy
2.3 H原子解离能
合金体系中H原子解离能的大小是衡量体系解氢能力的重要指标, H原子解离能的大小表示解离一个H原子所需吸收的热量, 即H原子解离能越小, 则合金体系的结构稳定性越差, 合金的解氢能力越强, 反之, 解氢能力越弱
[18 ]
。
式中, E- tot H 为超胞解离一个H原子后的总能量, Etot 为超胞的总能量, E (H 2) 为单个H分子的总能量。H原子的解氢能如表1所示, 单个H原子的解离能 (ΔE) 的大小顺序为:Mg6 H12 >Mg6 H11 F>Mg5 H12 Ni>Mg6 H12 F, 纯净的Mg6 H12 解离出单个H原子所需的能量为1.3953 eV, F, Ni合金化后解离出单个H原子所需的能量均小于纯净MgH2 的解氢能, 这说明F, Ni合金化后能够降低MgH2 体系的解氢能, 其中Mg6 H12 F解氢能力最强, Mg5 H12 Ni次之, Mg6 H11 F解氢能力较差。计算结果说明Ni, F元素能够改善MgH2 合金体系的解氢性能, 与Jin等
[9 ]
的实验结果一致。
2.4电子结构
通过总态密度 (DOS) 、分波态密度, 分析Mg H2 体系的解氢理论机制, 计算结果如图3所示, Mg6 H12 的总态密度 (DOS) 成键峰值主要分布在-45~-35 eV以及-10~10 eV区间, 在Feimi能级附近0~5 eV区间存在一个较宽的能隙;Ni置换Mg原子后Mg5 H12 Ni的成键峰值主要分布在-45~-35 eV以及-10~10 eV区间, 在-45~-35 eV区间成键峰值相对于Mg6 H12 明显降低并且峰值向低能级移动, 成键峰值由置换前的68.83eV降低为52.14 eV, 在Feimi能级附近Ni (d) , H (s) 的杂化形成峰值, 导致Feimi能级附近能隙减小, 费米能级附近能隙越宽, 则晶体结构的稳定性越高, 反之稳定性越低
[19 ]
, 说明Ni置换Mg原子后Mg5 H12 Ni的结构稳定性降低, 解氢难度降低。F置换H原子后Mg6 H11 F的总态密度相对于Mg6 H12 变化不明显, 低能级成键峰值为68.02 eV, 在-25~-20 eV区间形成一个较小的成键峰值主要是由F (s) 单独贡献, 在-10~0 eV区间F (p) , H (s) 杂化成键。F原子占据间隙位置后Mg6 H12 F的低能级成键峰值为51.18 eV, 成键峰值明显减小并且峰值明显向低能级移动, 在-30~-25 eV区间形成一个较小的成键峰值主要是由F (s) 单独贡献, 在Feimi能级附近F (p) , H (s) 的杂化成键, 导致Feimi能级附近能隙减小, MgH2 体系的稳定性变差, 解氢难度降低。
表1 单个H原子的解离能Table 1 Dissociation energy of a H atom (e V) 下载原图
表1 单个H原子的解离能Table 1 Dissociation energy of a H atom (e V)
MgH2 体系的交叠聚居数如表2所示, 纯净的Mg6 H12 第一邻近H-H键长为0.2502 nm, 第二邻近H-H键长为0.2768 nm;Mg6 H11 F超胞的第一、二邻近H-H键长变化不明显, 与态密度 (DOS) 分析结果一致, 说明Mg6 H11 F解氢效果不明显, MgF键长为0.2056 nm明显小于Mg-Mg键长0.2989nm;Mg6 H12 F超胞的第一邻近H-H键长为0.0828 nm, 与H2 分子的键长0.0741 nm非常接近, 聚居数为1.19, 聚居数为正说明成共价键, 数值越大共价键越强, 说明第一邻近H-H键极易形成H2 分子, F元素的加入促进了H2 分子的形成, 第二邻近H-H键长为0.2008 nm, Mg-F键长为0.2048 nm小于Mg-Mg键长0.2976 nm, 说明Mg与F元素容易成键形成更为稳定的化合物, 很好的解析了Mg6 H12 F形成热变大而解氢性能提高的反常现象;Mg5 H12 Ni超胞的第一邻近H-H键长为0.2343 nm, 第二邻近H-H键长为0.2399 nm, 相对于纯净的Mg6 H12 键长变短, Mg-Ni键长为0.2858 nm略大于Mg-Mg键长0.2849 nm说明Ni元素的加入形成不稳定的化合物。结合Jin等
[9 ]
的实验结果, 在MgH2 中加入Ni F2 球磨后出现Mg2 Ni H4 , 解氢反应完成后出现MgF2 。说明Mg与F元素容易成键形成更为稳定的MgF2 化合物, MgH2 与Ni元素形成不稳定的Mg2 Ni H4 化合物, F元素的加入促进了H2 分子的形成, 交叠聚居数分析结果从理论机制方面解析了Jin等
[9 ]
根据实验结果推测Ni F2 的加入促进了化学反应Ni F2 +3MgH2 =MgF2 +Mg2 Ni H4 +H2 的进行, 形成更为稳定的MgF2 和不稳定的Mg2 Ni H4 化合物。H-H键长变短, 形成H2 分子的倾向性变强
[6 ]
, Mg6 H12 F超胞与Mg5 H12 Ni超胞的第一、二邻近H-H键长明显变短, 从键长角度进一步解析了Mg5 H12 Ni, Mg6 H12 F改善MgH2 体系解氢性能的理论机制。
图3 MgH2合金的态密度Fig.3 Density states of MgH2alloy
(a) Mg6 H12 ; (b) Mg5 H12 Ni; (c) Mg6 H11 F; (d) Mg6 H12 F
表2 交叠聚居数Table 2 Overlap population 下载原图
表2 交叠聚居数Table 2 Overlap population
3结论
1.F原子很容易置换MgH2 中的H原子, 无论F原子占H位还是占居间隙位置都能形成更为稳定的结构。F原子占据间隙位置时, MgH2 体系解氢能力明显增加。
2.Ni原子置换Mg原子后, MgH2 体系的形成热与H原子的解离能都降低, 说明在MgH2 中加入Ni原子能够改善MgH2 体系的解氢性能。电子结构分析结果表明, MgH2 体系解氢能力的增加归因于H-H原子键长变短及Feimi能级附近的能隙变窄。
3.合金形成热以及交叠聚居数结果表明, Mg与F元素容易成键形成更为稳定的MgF2 化合物, Ni元素与MgH2 形成不稳定的Mg2 Ni H4 化合物, 从微观层面证实了Ni F2 的加入促进了化学反应Ni F2 +3MgH2 =MgF2 +Mg2 Ni H4 +H2 的进行, 从理论机制方面解析了Jin等
[9 ]
根据实验结果所作出的理论推测。
参考文献
[1] Ouyang L Z, Cao Z J, Wang H, Hu R Z, Zhu M. Application of dielectric barrier discharge plasma-assisted milling in energy storage materials―A review[J]. J.Alloys Compd., 2017, (691) :422.
[2] Ismail M. Effect of La Cl3addition on the hydrogen storage properties of MgH2[J]. Energy, 2015, 79 (1) :177.
[3] Song Y, Guo Z X, Yang R. Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications MgH2[J]. Physical Review Letters B, 2014, 12 (3) :132106.
[4] Zaluski L, Zaluska A, Strom-Olsen J O. Hydrogen absorption in nanocrystalline Mg2Ni formed by mechanical alloying[J]. J. Alloys Compd., 1995, 217:24.
[5] Suda S. Studies on the fluorination method for improving surface properties and characteristics of AB5-typesof hydrides[J]. J. Alloys Compd., 2002, 330:627.
[6] Zhou D W, Zhang J, Liu J S. Mechanism of dehydrogenating properties of Ni doped MgH2systems[J]. Chinese Journal of Nonferrous Metals, 2009, 19 (2) :315. (周惦武, 张健, 刘金水. Ni掺杂MgH2体系解氢性能的机制[J].中国有色金属学报, 2009, 19 (2) :315.)
[7] Yuan J, Zhou D W, Wei H W. First-principles investigation of Ti F3solution hydrogen thermodynamic effects on MgH2[J]. Chinese Journal of Nonferrous Metals, 2016, 26 (7) :1480. (袁江, 周惦武, 魏红伟. Ti F3对MgH2体系解氢热力学影响的第一性原理研究[J].中国有色金属学报, 2016, 26 (7) :1480.)
[8] Zhang J, Tang W, Shao L, Yu X F, Long C G, Chen J.Microstructures and dehydrogenation properties of ballmilled MgH2-K2Ti6O13-Ni composite systems[J]. Journal of Materials Engineering, 2016, 44 (11) :101. (张健, 汤旺, 邵磊, 余小峰, 龙春光, 陈荐. MgH2-K2Ti6O13-Ni球磨复合体系的微观结构与解氢性能[J].材料工程, 2016, 44 (11) :101.)
[9] Jin S A, Shim J H, Cho Y W, Yi K W. Dehydrogenation and hydrogenation characteristics of MgH2with transition metal fluorides[J]. J. Power Sources, 2007, 172 (1/2) :859.
[10] Liu Y C, Gao L J. Effects of Co concentration on the mechanical properties of Ni Al[J]. Rare Metals and Cemented Carbides, 2013, 41 (6) :33. (刘友成, 高丽洁.合金化元素Co浓度对Ni Al力学性能的影响[J].稀有金属与硬质合金, 2013, 41 (6) :33.)
[11] Lindan P L D, Segall M D, Probert M J. First-principles simulation:ideas illustrations and the CASTEP code[J]. Phys:Condens. Matter, 2002, 14 (11) :2717.
[12] Miwa K, Ohba N, Towata S I, Nakamori Y, Orimo S I.First-principles study on lithium borohydride Li BH4[J].Phys. Rev. B, 2004, 69:245120.
[13] Li C, Zhou D W, Peng P, Wan L. First-principles calculation on dehydrogenating properties of Li BH4-X (X=O, F, Cl) systems[J]. Acta Chimica Sinica, 2012, 70 (1) :71. (李闯, 周惦武, 彭平, 万隆. Li BH4-X (X=O, F和Cl) 体系解氢性能的第一原理计算[J].化学学报, 2012, 70 (1) :71.)
[14] Yin L C, Wang P, Fang Z Z, Cheng H M. Thermodynamically tuning Li BH4by fluorine anion doping for hydrogen storage:a density functional study[J]. Chem.Phys. Lett., 2008, 450:318.
[15] Nakamura H, Nguyen-Manh D, Pettifor D G. Electronic structure and energetics of La Ni5, α-La2Ni10H andβ-La2Ni10H14[J]. J. Alloys Compd., 1998, 281:81.
[16] Zhou D W, Peng P, Liu J S. First-principles calculation of hydrogenating properties of MgH2-V systems[J].Science in China Series E, 2006, 49 (2) :129.
[17] Bogdanovic'a B, Bohmhammelb K, Christb B, Reisera A, Schlichtea K, Vehlena R, Wolfb U. Thermodynamic investigation of the magnesium-hydrogen system[J]. J. Alloys Compd., 1999, 282:84.
[18] Xia L S, Zhu S H. Mechanism of dehydrogenation of Al-LiBH4systems[J]. Chinese Journal of Rare Metals, 2013, 37 (7) :531. (夏罗生, 朱树红. Al-LiBH4体系解氢性能的机制研究[J].稀有金属, 2013, 37 (7) :531.)
[19] Wang J, Wang G, Zhao J. Density-functional study of Aun (n=2~20) clusters:lowest-energy structures and electronic properties[J]. Phys. Rev. B, 2002, 66:035418.