
Potential energies of characteristic atoms on basis of experimental heats of formation of AuCu and AuCu3 compounds (Ⅰ)
XIE You-qing(谢佑卿)1, 2, 3, LIU Xin-bi(刘心笔)1, 2, 3, LI Xiao-bo(李晓波)4,
PENG Hong-jian(彭红建)1, 2, 3, NIE Yao-zhuang(聂耀庄)1, 2, 3
1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Powder Metallurgy Research Institute, Central South University, Changsha 410083, China;
3. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China;
4. College of Mechanical Engineering, Xiangtan University, Xiangtan 411105, China
Received 30 March 2009; accepted 2 June 2009
Abstract: The systematic science of alloys(SSA) is a framework of the total energy and total volume able to be separated. The potential energy sequences of characteristic atoms at the central sites of the basic clusters in the fcc-based lattice Au-Cu system are separated out from smaller experimental heats of formation of L10-AuCu and L12-AuCu3 compounds only, by nine potential energy E-functions and through the use of structural unit inversion method. From these potential energy sequences, the potential energies and heats of formation of the disordered Au1-xCux alloys at 0 K are calculated. The potential energies, heats of formation and Tc-temperatures of order-disorder transitions of the L10-AuCu, L12-Au3Cu and L12-AuCu3 compounds, as well as the Au3Cu-, AuCu- and AuCu3- type ordered alloys with maximal ordering degrees are calculated too. The results show that the 5th E-function may be chosen for developing it into the free energy-, enthalpy-, vibrational energy- and vibrational entropy-functions for describing thermodynamic properties of the compounds, ordered and disordered phases and for establishing the phase diagram of the Au-Cu system in the future.
Key words: systematic science of alloys; Au-Cu system; potential energy; heat of formation; order-disorder transition temperature
1 Introduction
An alloy system contains three structure levels: the phase level of organizations, atomic level of phases and electronic level of atoms. In order to get a entirely understanding of the alloy systems, to establish phase diagrams and to search a method for designing of alloys, the SSA framework[1-4] has been established based on two scientific philosophy propositions. A diversity of structures, properties and features of whether matter or nonmatter systems should be attributed to combination and arrangement of structural units in the structural unit sequence. For examples, the diversity of atoms in the atomic system is attributed to arrangement of electrons in the electronic orbital (state) sequence described by four quantum numbers, which are the main quantum number, angular quantum number, magnetic quantum number and spin quantum, under the influence of nuclei; the diversity of substances in the matter system is attributed to compositions of elements in the periodic sequence of elements; the diversity of quantities in metrology is described by combination and arrangement of basic numbers in the basic number sequence, which are Arabic numerals; the diversity of species in the biological system is attributed to splices of the genes in the gene sequence; and the diversity of individual characters in character system is described by combination and arrangement of stroke elements in the stroke element sequence, which are the dot, horizontal, vertical, left-falling, right-falling, turning and hook strokes. And a systematic theory of any complex system described quantitatively should be constructed by structural unit sequence-, equation- and information-chains. Besides the quantum-mechanics of free atom system, there is few or no one described quantitatively. The SSA framework is constructed indeed by the structural unit sequence-, equation- and information-chains.
1.1 Models and structure unit sequence chain
The atomic level of alloy phases in the SSA framework involves three models for constructing diversity of structures and properties of alloy phases [5-8].
1) The basic cluster overlapping(BCO) model, of which the structural units are a pair of basic cluster sequences in a based lattice (such as fcc, hcp and bcc) of binary alloy systems, with each cluster consisting of a central atom, the first neighbor configuration, the second neighbor configuration and the third neighbor configuration. The actions of the BCO model are to give information about an atomic arrangement of alloy phases described by overlapping pattern of basic clusters and to determine the splitting order on the potential energy, volume and electronic structure of the central atoms, which may have one, two and three splitting orders respectively corresponding to the basic clusters with one, two and three neighbor configurations. Using one order split the basic consists of a central atom and the first neighboring configuration [(I-i)Au, iCu], which contains i Cu-atoms and (I-i) Au-atoms. Here the symbol α denotes Au or Cu; I is the coordinative number and equals 12 for the fcc based lattice; and i can change from 0 to 12.
2) The characteristic atom arranging(CAA) model, of which the structural units are a pair of characteristic atom sequences, with each characteristic atom being the central atom of a specific basic cluster. The characteristic 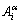 atom has own characters at the ground state: potential energy
atom has own characters at the ground state: potential energy 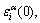 volume
volume 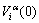 and electronic structure
and electronic structure  The actions of the CAA model are to give information about an atomic arrangement of alloy phases described by potential energy pattern, volume pattern and electronic structure pattern of the characteristic atoms occupied at the various lattice sites in intermetallics[5-8], to derive equations for calculating concentrations of various characteristic atoms and configurational entropy of ordered and disordered alloys as functions of composition(x) and ordering degree(σ) and to establish composition-ordering degree dependent E(x, 0, σ), V(x, 0, σ) and ψ(x, 0, σ) functions of potential energy, volume and electronic structure of alloy phases at 0 K or without considering temperature effect.
The actions of the CAA model are to give information about an atomic arrangement of alloy phases described by potential energy pattern, volume pattern and electronic structure pattern of the characteristic atoms occupied at the various lattice sites in intermetallics[5-8], to derive equations for calculating concentrations of various characteristic atoms and configurational entropy of ordered and disordered alloys as functions of composition(x) and ordering degree(σ) and to establish composition-ordering degree dependent E(x, 0, σ), V(x, 0, σ) and ψ(x, 0, σ) functions of potential energy, volume and electronic structure of alloy phases at 0 K or without considering temperature effect.
Therefore, in a based lattice the variations in potential energy, volume and electronic structure of intermetallics, various type ordered alloys and disordered alloys with composition and ordering degree can be calculated by the same information about a pair of potential energy sequences, a pair of volume sequences and a pair of electronic structure sequences, which belong to a pair of characteristic atom sequences.
3) The characteristic crystal mixing(CCM) model, of which the structure units are a pair of invented characteristic crystal sequences, with each characteristic crystal consisting of the same characteristic atoms of the identical potential energy, identical volume and identical electronic structure. The actions of the CCM model are to establish a set of temperature(T) dependent functions of the energetic and volumetric properties consisting of the general thermal expansion coefficient βv(T) function, general thermal expansion volume Vv(T) function, general vibrational capacity  function, general vibrational energy Uv(T) function, general vibrational entropy Sv(T) function, enthalpy H(T) function and free energy G(T) function of the characteristic crystals and to derive a set of composition-temperature-ordering degree dependent functions of the the general thermal expansion coefficient βv(x, T, σ), general thermal expansion volume Vv(x, T, σ), general vibration heat capacity
function, general vibrational energy Uv(T) function, general vibrational entropy Sv(T) function, enthalpy H(T) function and free energy G(T) function of the characteristic crystals and to derive a set of composition-temperature-ordering degree dependent functions of the the general thermal expansion coefficient βv(x, T, σ), general thermal expansion volume Vv(x, T, σ), general vibration heat capacity 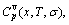 general vibrational energy Uv(x, T, σ), general vibration entropy Sv(x, T, σ), enthalpy H(x, T, σ) and characteristic free energy G*(x, T, σ) without containing configuration entropy Sc(x, σ) of alloy phases. And finally, by combining the G*(x, T, σ) function with partition function, the free energy G(x, T, σ) function can be derived, where the G(x, T, σ) function contains configuration entropy Sc(x, σ).
general vibrational energy Uv(x, T, σ), general vibration entropy Sv(x, T, σ), enthalpy H(x, T, σ) and characteristic free energy G*(x, T, σ) without containing configuration entropy Sc(x, σ) of alloy phases. And finally, by combining the G*(x, T, σ) function with partition function, the free energy G(x, T, σ) function can be derived, where the G(x, T, σ) function contains configuration entropy Sc(x, σ).
These three models are proposed in order to overcome disappointments of the atomic pair interaction model and central atom model[9] as well as effective cluster interaction model[10-12].
1.2 Equation and information chains
1.2.1 Additive law of q-properties of characteristic crystals
According to CCM model, the extensive properties q(x, T, σ), qA(x, T, σ), qB(x, T, σ) of a given alloy phase and its components can be obtained by an additive law of the q-properties of characteristic crystals (in terms of CCA law)[5-8]:
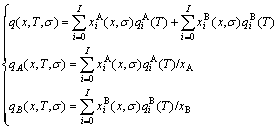 (1)
(1)
where  and
and  are the concentrations of the characteristic atoms.
are the concentrations of the characteristic atoms.
1.2.2 Nine E(x, 0, σ)- and nine V(x, 0, σ)-functions of alloy phases at 0 K
In order to make Eq.(1) become a simple, applicable and separable q(x, 0, σ) function (here q denotes E or V), three type relations of 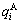 and
and 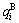 with i have been designed[1, 8]:
with i have been designed[1, 8]:
TypeⅠof straight line relation:
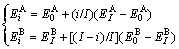 (2)
(2)
Type Ⅱ of concave parabola relation:
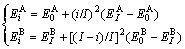 (3)
(3)
Type Ⅲ of convex parabola relation:
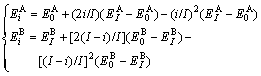 (4)
(4)
where  and
and 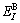 denote, respectively, potential energies of the primary
denote, respectively, potential energies of the primary  and
and 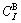 characteristic crystals;
characteristic crystals; 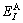 and
and 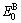 denote, respectively, potential energies of the terminal
denote, respectively, potential energies of the terminal  and
and  characteristic crystals.
characteristic crystals.
By combining Eqs.(2), (3), (4) and substituting them into Eq.(1), nine E(x, 0, σ) functions can be obtained. In Table 1 the nine general E(x, 0, σ)-functions of alloy phases can be used to compounds, ordered and disordered alloy phases. In Table 2, the nine E(x, 0, 0)- functions can be used to the disordered alloy phase only.
Table 1 Nine general potential energy E(x, 0, σ)-functions of alloy phases at 0 K
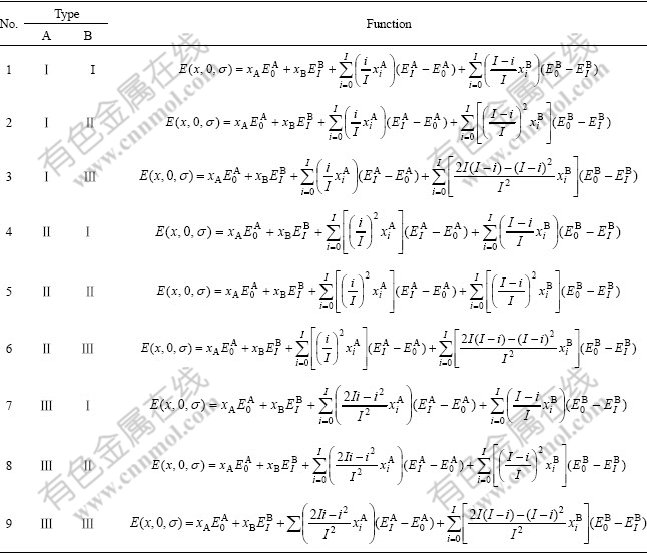
Table 2 Nine potential energy E(x, 0, 0)-functions of disordered alloy phases at 0 K

1.3 Methodology
From the first proposition, an inference can be drawn: “The whole can be reproduced from parts”. For example, the whole of a tree can be reproduced from a seed, a leaf or a branch of the tree in biologic systems; The whole information of an alloy system can be reproduced from disordered alloys, ordered alloys or intermetallics. This means that “The total potential energies and total volumes of a few alloys can be separated into the potential energy sequence and volume sequence of the characteristic crystals (atoms), from which the whole information about energetic and volumetric properties, electronic and crystalline structures of all alloy phases can be reproduced”. Therefore, the systematic study for an alloy system in the SSA framework is divided into three step investigations.
1) The aims of the first step investigation are to choose E(x, 0, σ) and V(x, 0, σ) functions and to determine a pair of potential energy sequences and a pair of volume sequences of characteristic crystals (atoms), through resolving nine E(x, 0, σ)- and V(x, 0, σ)- functions on the basis of FP-calculated heats and volumes of formation of several intermetallics only, or of experimental heats and volumes of formation of several intermetallics only and several disordered alloys only by the structural unit inversion method; to determine a pair of electronic structure sequences of characteristic crystals (atoms), according to potential energies and volumes of characteristic crystals (atoms), then to study energetic properties, volumetric properties and electronic structures of alloy phases at ground state (0 K).
2) The aims of the second step investigation are to obtain energetic and volumetric properties of characteristic crystals as function of temperature, to obtain the energetic and volumetric property equations of alloy phases as functions of composition, temperature and ordering degree and then to study energetic and volumetric properties, crystalline parameters and electronic structures of alloy phases in the whole ranges of composition, temperature and ordering degree.
3) The aims of the third step investigation are to study stability, transformation and equilibrium of alloy phases, to establish phase diagram and to provide a systematic knowledge about energetic and volumetric and structural properties, crystalline and electronic structures of characteristic crystal (atom) sequences for designing applied alloys. That is called as design engineering of characteristic crystal (atom) sequences of alloy systems.
The Au-Cu system has a diversity of alloy phases, which include intermetallics with various space groups, ordered alloys with Au3Cu-, AuCu-, and AuCu3-types and disordered alloys, a relative simplicity for systematical study, which indicates all solid phases exist in the fcc-based lattice only, and is rich in order-disorder phenomena with phase transitions at Au3Cu, AuCu and AuCu3, that leads to the fact that it has become a studying platform for nearly all theories and experimental techniques of alloys. Now, it is also used as the platform systematically to represent the SSA framework through a series of works. The outline of remainder of this work is as follows. In the next section, the results are presented. The potential energy sequences of the  and
and 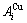 characteristic atoms are calculated by nine E-functions at 0 K; The potential energies and heats of formation of disordered Au1-xCux alloys are calculated by nine E-functions at 0 K; The potential energies and heats of formation and critical Tc-temperatures of the order-disorder transformation of Au3Cu, AuCu and AuCu3 compounds are calculated by nine E-functions; The potential energies and heats of formation and critical Tc-temperatures of the order-disorder transformation of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys, and some general conclusions are then presented.
characteristic atoms are calculated by nine E-functions at 0 K; The potential energies and heats of formation of disordered Au1-xCux alloys are calculated by nine E-functions at 0 K; The potential energies and heats of formation and critical Tc-temperatures of the order-disorder transformation of Au3Cu, AuCu and AuCu3 compounds are calculated by nine E-functions; The potential energies and heats of formation and critical Tc-temperatures of the order-disorder transformation of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys, and some general conclusions are then presented.
2 Results
2.1 Potential energy sequences of characteristic atoms
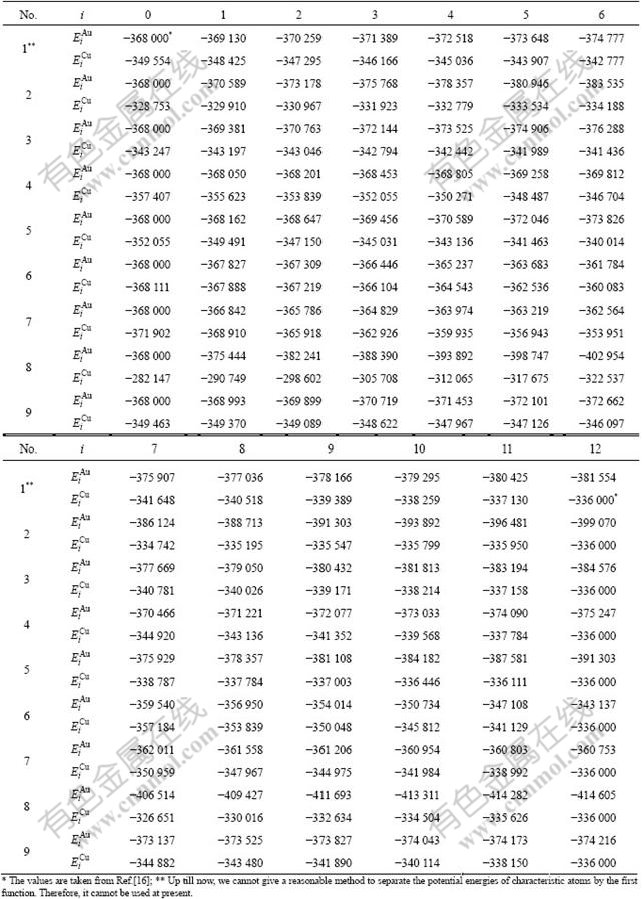
The experimental data show that from 0 K to 560 K for AuCu compound and from 0 K to 400 K for AuCu3 compound, their heats of formation have no variations with temperature[13]. Therefore, it can be considered that the ?Hexp(AuCu, 298, 1)=?E(AuCu, 0, 1)=-8 745 J/mol, and ?Hexp(AuCu3, 298, 1)=?E(AuCu3, 0, 1)= -7 164 J/mol[13], which are smaller respectively than ?Hexp(AuCu, 298, 1)=-9 337 J/mol and ?Hexp(AuCu3, 298, 1)=-7 268J /mol[14-15]. From the smaller experimental heats of formation, the potential energy 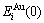 and
and 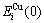 sequences of the and characteristic atoms in the Au-Cu system separated out by nine E-functions in Table1 are listed in Table 3 and shown in Fig.1. From these results, it can be known that the potential energy sequences obtained from nine E-functions are different and that the and sequences obtained by the 4th, 5th and 7th E-functions are different too, even the average potential energies of the compounds, ordered and disordered alloys calculated by them are, respectively, equivalent. The 2nd and 3rd E-functions have the same characters (see results of 2.2, 2.3 and 2.4 in the present section).
sequences of the and characteristic atoms in the Au-Cu system separated out by nine E-functions in Table1 are listed in Table 3 and shown in Fig.1. From these results, it can be known that the potential energy sequences obtained from nine E-functions are different and that the and sequences obtained by the 4th, 5th and 7th E-functions are different too, even the average potential energies of the compounds, ordered and disordered alloys calculated by them are, respectively, equivalent. The 2nd and 3rd E-functions have the same characters (see results of 2.2, 2.3 and 2.4 in the present section).
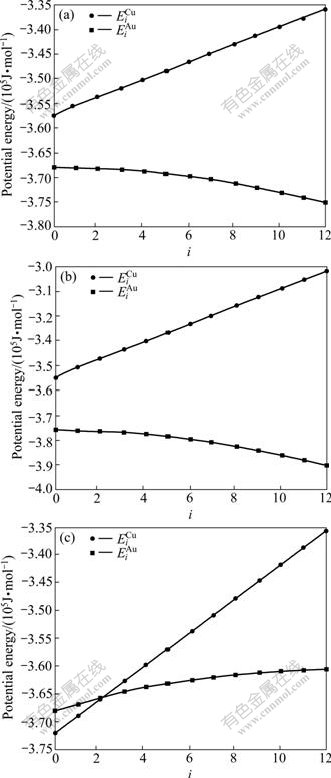
Fig.1 Potential energy 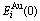 and
and 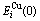 sequences of
sequences of 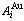 and
and  characteristic atoms in Au-Cu system obtained by the 4th (a), 5th (b) and 7th (c) E-functions
characteristic atoms in Au-Cu system obtained by the 4th (a), 5th (b) and 7th (c) E-functions
Table 3 Potential energy sequences of 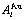 and characteristic atoms in Au-Cu system calculated by nine E-functions at 0 K (J/mol)
and characteristic atoms in Au-Cu system calculated by nine E-functions at 0 K (J/mol)
2.2 Energetic properties of disordered Au1-xCux alloys and their components
Fig.2 shows the distribution of the 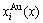 and
and  of characteristic atoms in the disordered Au1-xCux alloys.
of characteristic atoms in the disordered Au1-xCux alloys.
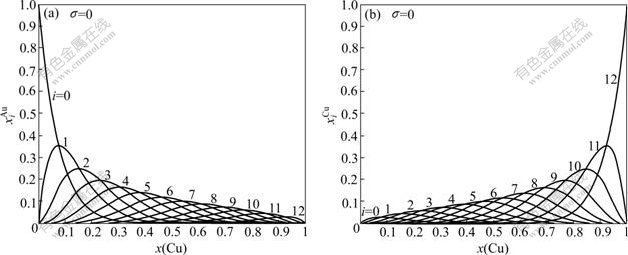
Fig.2 Concentrations  and
and 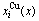 in disordered Au1-xCux alloys as functions of composition x(Cu)
in disordered Au1-xCux alloys as functions of composition x(Cu)
According to the potential energy sequences of the characteristic atoms in Table 3, the E(x, 0, 0), EAu(x, 0, 0) and ECu(x, 0, 0) potential energies of disordered Au1-xCux alloys and their components as well as their ?H(x, 0, 0) heats of formation calculated by nine E-functions in Table 2 are listed in Table 4 and shown in Fig.3. From these results, the following knowledge can be obtained.
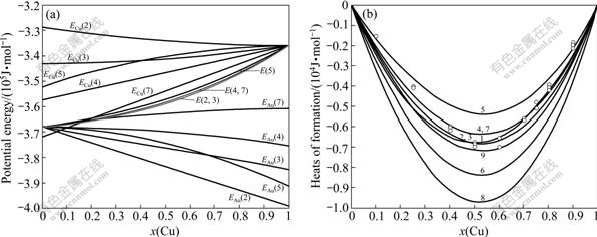
Fig.3 Average potential energies E(x, 0, 0), EAu(x, 0, 0) and ECu(x, 0, 0) of disordered Au1-xCux alloys and their components calculated from the 2nd, 3rd, 4th, 5th and 7th E-functions (a) and ?H(x, 0, 0) for the disordered Au1-xCux alloys calculated from nine E-functions, together with experimental heats of formation at 320 K[15] (denoted by circles) (b)
Table 4 Potential energies E(x, 0, 0), EAu(x, 0, 0), ECu(x, 0, 0) and heats of formation ?H(x, 0, 0) of disordered Au1-xCux alloys calculated by nine E-functions at 0 K (J/mol)
1) The average E(x, 0, 0) potential energies of the disordered Au1-xCux alloys obtained from nine E-functions are different each other. The average EAu(x, 0, 0) and ECu(x, 0, 0) potential energies of the Au and Cu components obtained by nine E-functions are different each other too. But there is no E-function, which can well describe the experimental heats of formation in the whole compositional range. It has been discovered that the transformation of the disordered Au0.5Cu0.5 alloy to ordered AuCu alloy is fast at temperatures below 573 K so that it is rather difficult to fully suppress ordering on quenching[17-20]. Therefore, the experimental heats of formation of so-called disordered alloys in the compositional range (atomic fraction) 25%≤x(Cu)≤75% are referred to partly-ordered alloys with short- range ordering degrees. The details will be reported in the second part of this investigation.
2) The average potential energies and heats of formation of the disordered Au1-xCux alloys obtained by the 5th E-functions are higher than corresponding experimental values. It may be chosen to describe the Au-Cu system, considering that the experimental heats of formation of the so-called disordered alloys are referred to those of partly-ordered alloys with short-range ordering degrees.
2.3 Energetic properties of L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds
In the SSA framework, the crystalline structures of compounds are described by the characteristic atom occupation pattern, in which one can distinguish characteristic atoms occupied at the lattice sites (see Fig.4).
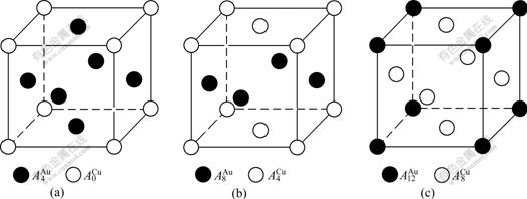
Fig.4 Characteristic atom occupation patterns of L12-Au3Cu (a), L10-AuCu (b) and L12-AuCu3 (c) crystalline structures
According to the potential energy sequences of the characteristic atoms in the fcc-based Au-Cu system, the potential energies  and
and  of characteristic atoms occupied at the ith sites in the cell, the average potential energies E(x, 0, 0) and heats of formation ?H(x, 0, 0) of the compounds can be calculated by CCA law. If the vibrational contribution to and the effect of variation in ordering degree on the entropy and enthalpy are neglected, the critical Tc(0)-temperature of the order-disorder phase transformation is calculated by the potential energy difference divided by the congfigurational entropy difference:
of characteristic atoms occupied at the ith sites in the cell, the average potential energies E(x, 0, 0) and heats of formation ?H(x, 0, 0) of the compounds can be calculated by CCA law. If the vibrational contribution to and the effect of variation in ordering degree on the entropy and enthalpy are neglected, the critical Tc(0)-temperature of the order-disorder phase transformation is calculated by the potential energy difference divided by the congfigurational entropy difference:
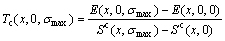 (10)
(10)
The results are listed in Table 5 and shown in Fig.5, from which the following knowledges can be obtained:
Table 5 Potential energies of characteristic atoms (ε, in 10-19 J/atom), total potential energies (E, in J/mol), heats of formation (?H, in J/mol) and order-disorder transformation temperatures (Tc, in K) of L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds at 0 K

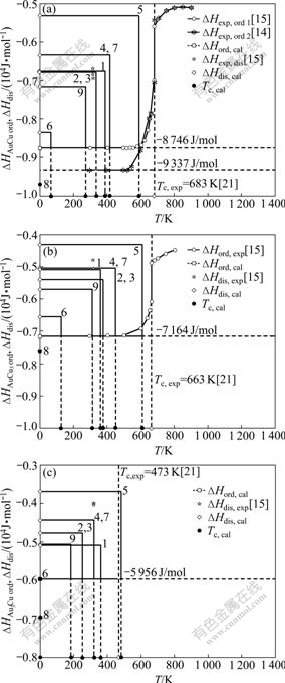
Fig.5 ?Hord(0)-?Hdis(0)―Tc interrelated patterns of AuCu (a), AuCu3 (b) and Au3Cu (c) compounds relative to corresponding disordered Au1-xCux alloys calculated from nine E-functions together with experimental data
1) The average potential energies of these compounds obtained from the 2nd to 9th E-functions are equal each to each, but their potential energies of characteristic atoms are different each to each.
2) The average potential energies and heats of formation of the L10-AuCu and L12-AuCu3 compound obtained by the 2nd to 9th E-functions are equal respectively to corresponding experimental values, but only the Tc-temperatures obtained by 5th E-function are closer by experiment values than ones obtained by other E-functions.
3) The average potential energies and heats of formation of the L12-Au3Cu compound calculated from the 2nd to 9th E-functions are equal each to each and closer by the experiment values, but only the Tc-temperature obtained by the 5th E-function is closer by experiment value than ones obtained by other E-functions.
4) The ?Hord(0)-?Hdis(0)―Tc(0) interrelated patterns of the AuCu, AuCu3 and Au3Cu compounds relative to corresponding disordered alloys obtained respectively from nine E-functions show that the heats of formation and Tc-temperatures calculated by 5th E-function can be satisfactory for describing experimental values at the same time.
2.4 Energetic properties of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys
In the SSA framework, the ordering of disordered alloys is attributed to degeneracy in the energy, volume and electronic structure states of characteristic atoms, and the disordering of ordered alloys is attributed to split in the energy, volume and electronic structure states of characteristic atoms. According to the potential energy sequences and concentrations 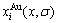 and
and 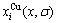 in the ordered Au1-xCux alloys with ordering degree σ, their energetic properties can be calculated by nine E-functions in Table 1. The concentrations
in the ordered Au1-xCux alloys with ordering degree σ, their energetic properties can be calculated by nine E-functions in Table 1. The concentrations 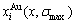 and
and 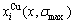 of characteristic atoms and some energetic properties of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees are shown in Figs.6 and 7, but only the calculated energetic properties of the Au-Cu-type ordered alloys are listed in Table 6.
of characteristic atoms and some energetic properties of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees are shown in Figs.6 and 7, but only the calculated energetic properties of the Au-Cu-type ordered alloys are listed in Table 6.
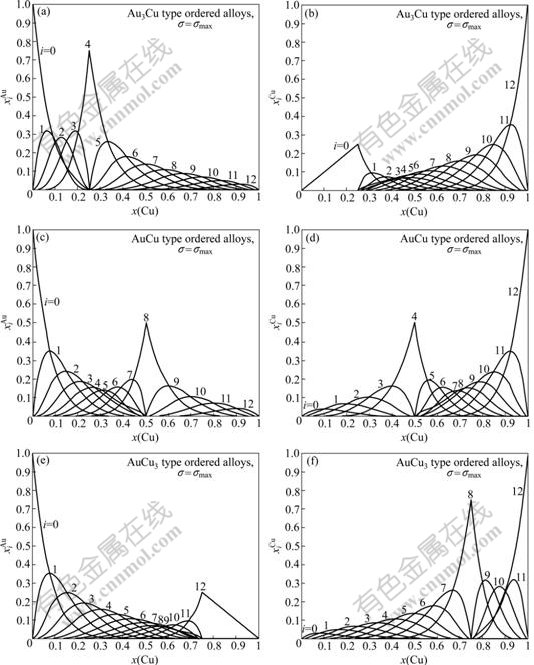
Fig.6 Concentrations  and
and  of characteristic atoms in Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees
of characteristic atoms in Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees
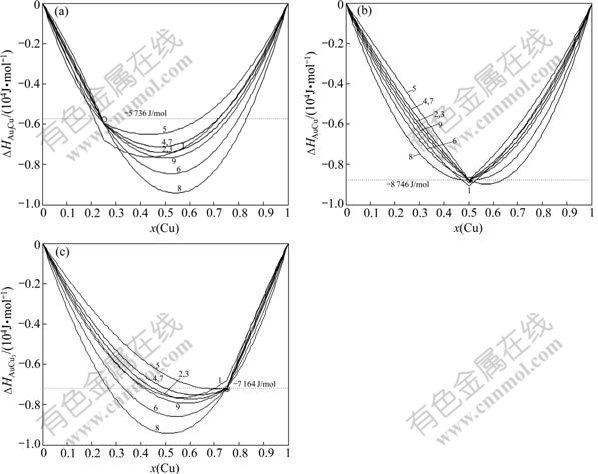
Fig.7 Calculated heats of formation 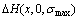 of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees by nine E-functions at 0 K (Experimental values[13] denoted by circles are respectively: ?H(Au3Cu) =-5 736 J/mol, ?H(AuCu)=-8 746 J/mol, ?H(AuCu3)=-7 164 J/mol
of Au3Cu-, AuCu- and AuCu3-type ordered Au1-xCux alloys with maximal ordering degrees by nine E-functions at 0 K (Experimental values[13] denoted by circles are respectively: ?H(Au3Cu) =-5 736 J/mol, ?H(AuCu)=-8 746 J/mol, ?H(AuCu3)=-7 164 J/mol
Table 6 Potential energies  and heats of formation of AuCu-type ordered Au1-xCux alloys with maximal ordering degree calculated by nine E-functions at 0 K (J/mol)
and heats of formation of AuCu-type ordered Au1-xCux alloys with maximal ordering degree calculated by nine E-functions at 0 K (J/mol)
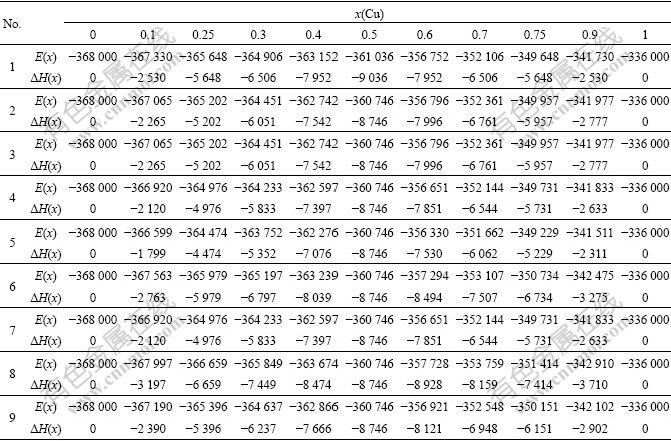
According to Eq.(10), the critical temperatures Tc(x, 0, σmax) of Au3Cu-, AuCu- and AuCu3-type Au1-xCux alloys translated into the disordered alloys described by nine E-functions have been calculated. The Tc(0)-temperatures obtained from 5th E-function are closer respectively by the experimental values than from other E-functions (see Fig.8).
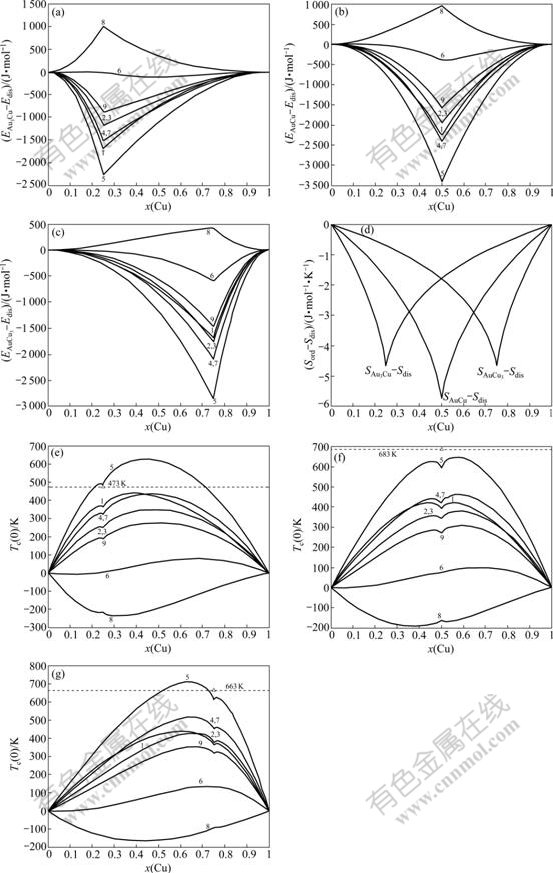
Fig.8 Potential energy differences (a, b, c), configurational entropies 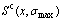 differences (d) and critical temperatures (e, f, g) of Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximum ordering degrees relative to disordered Au1-xCux alloys
differences (d) and critical temperatures (e, f, g) of Au3Cu-, AuCu- and AuCu3-type ordered alloys with maximum ordering degrees relative to disordered Au1-xCux alloys
3 Conclusions
1) From the smaller experimental heats of formation of the L10-AuCu and L12-AuCu3 compounds, only the potential energy sequences have been separated out by nine E-functions. Although the potential energies of the L12-Au3Cu, L10-AuCu and L12-AuCu3 compounds calculated from the 2nd to 9th E-functions are equal each to each, the potential energies of their characteristic atoms are not equal each to each.
2) According to the potential energy sequences of the nine E-functions, the heats of formation of the disordered Au1-xCux alloys have been calculated. But there is no E-function, which can well describe the experimental heats of formation in the whole compositional range. It has been discovered that the so-called experimental disordered Au1-xCux alloys are the partly ordered alloys with short range ordering degree, which will be reported in the second part of this investigation.
3) The Tc(0) -temperatures of the Au3Cu, AuCu and AuCu3 compounds obtained from the 5th E-function are closer by the experimental values than from other E-functions. The Tc(0)-temperatures of these three compounds obtained from the other E-functions are greatly lower than experimental ones.
4) The 5th E-function may be chosen for developing it into the free energy, enthalpy, vibrational energy and vibrational entropy functions for describing the composition- temperature-ordering degree dependence of thermodynamic properties of the compounds, ordered and disordered phases, and for establishing the phase diagram of the Au-Cu system in the future.
5) Although the potential energy sequences of characteristic atoms are separated out from the experimental heats of formation of both L10-AuCu and L12-AuCu3 compounds only, which can be used for calculating energetic properties of compounds, ordered and disordered alloys. This demonstrates that the first scientific philosophy proposition about the diversity of structures and properties of the system is correct, and that the whole energetic information can be reproduced from a part energetic information.
References
[1] XIE You-qing. Atomic energies and Gibbs energy functions of Ag-Cu alloys [J]. Science in China (Series E), 1998, 41: 146-156.
[2] XIE You-qing, ZHANG Xiao-dong. Atomic volumes and volumes functions of Ag-Cu alloys [J]. Science in China (Series E), 1998, 41: 157-168.
[3] XIE You-qing, ZHANG Xiao-dong. Electronic structure of Ag-Cu alloys [J]. Science in China (Series E), 1998, 41: 225-236.
[4] XIE You-qing, ZHANG Xiao-dong. Phase diagram and thermodynamic properties of Ag-Cu alloys [J]. Science in China (Series E), 1998, 41: 348-356.
[5] XIE You-qing, TAO Hui-jing, PENG Hong-jian, LI Xiao-bo, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC TiAl2 type alloys [J]. Physica B, 2005, 366: 17-37.
[6] XIE You-qing, PENG Hong-jian, LIU Xin-bi, PENG Kun. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC Ti3Al-type alloys [J]. Physica B, 2005, 362: 1-17.
[7] XIE You-qing, LIU Xin-bi, PENG Kun, PENG Hong-jian. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC TiAl3-type alloys [J]. Physica B, 2004, 353: 15-33.
[8] XIE You-qing, PENG Kun, LIU Xin-bi. Influences of xTi/xAl on atomic states, lattice constants and potential energy planes of ordered FCC TiAl-type alloys [J]. Physica B, 2004, 344: 5-20.
[9] LUPIS C P P. Chemical thermodynamics of materials [M]. Amsterdam: North-Holland, 1983: 438-469.
[10] ASTA M, WOLVERTON C, de FONTAINE D, DREYSS? H. Effective cluster interactions from cluster-variation formalism (I) [J]. Phys Rev B, 1991, 44: 4907-4913.
[11] ASTA M, WOLVERTON C, de FONTAINE D, DREYSS? H. Effective cluster interactions from cluster-variation formalism (II) [J]. Phys Rev B, 1991, 44: 4913-4924.
[12] OZOLIN? V, WOLVERTON C, ZUNGER A. Cu-Au, Ag-Au, Cu-Ag and Ni-Au intermetallics: First-principles study of temperature-composition phase diagrams and structure [J]. Phys Rev B, 1998, 57: 6427-6442.
[13] HULTGREN R, DESAI P D, HAWKINS D T, GLEISER M, KELLEY K K. Selected values of thermodynamic properties of binary metals and alloys [M]. Metal Park, OH: American Society for Metals, 1973.
[14] ORR R L, LUCIAT-LABRY J, HULTGREN R. Energy of the order-disorder transformation in Au-Cu [J]. Acta Metallurgica, 1960, 8: 431-434.
[15] ORR R L. Heats of formation of solid Au-Cu alloys [J]. Acta Metallurgica, 1960, 8: 489-493.
[16] KITTEL C. Solid state physics [M]. 6th Ed. New York: Wiley, 1995.
[17] van TENDELOO G, AMELINCKX S, JENG S J, WAYMAN C M. The initial stages of ordering in CuAu (I) and CuAu (II) [J]. J Mater Sci, 1986, 21: 4395-4402.
[18] BATTEZZATI L, BELOTTI M, BRUNELLA V. Calorimetry of ordering and disordering in AuCu alloys [J]. Scripta Mater, 2001, 44: 2759-2764.
[19] XIE You-qing, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Potential energy sequences of characteristic atoms on basis of heats of formation of disordered Au1-xCux alloys (I) [J]. Journal of Materials Science and Engineering, 2009, 3: 51-68.
[20] XIE You-qing, LI Xiao-bo, LIU Xin-bi, PENG Hong-jian, NIE Yao-zhuang. Potential energy sequences of characteristic atoms on basis of heats of formation of disordered Au1-xCux alloys (II) [J]. Journal of Materials Science and Engineering, 2009, 6: 44-57.
[21] BARRETT C S, MASSALSKI T B. Structure of metals [M]. 3rd ed. New York: McGraw-Hill, 1966.
Foundation item: Project(50471058) supported by the National Natural Science Foundation of China; Project(08JJ3099) supported by the Natural Science Foundation of Hunan Province, China
Corresponding author: XIE you-qing; Tel: +86-731-88879287; E-mail: xieyouq2000@yahoo.com.cn
DOI: 10.1016/S1003-6326(08)60436-7
(Edited by YANG Hua)

