单原子状态自洽法的算法
彭红建1, 2,谢佑卿2,李小波2
(1. 中南大学 化学化工学院,湖南 长沙,410083;
2. 中南大学 材料科学与工程学院,湖南 长沙,410083)
摘 要:针对传统的算法存在解的精度不高和计算时间长等问题,通过对单原子状态自洽法的算法进行分析,利用数值运算功能极强的MATLAB(MATrix LABoratory)为工具,以金属Cu为例,对确定晶体电子结构的计算方法单原子状态自洽法编程求解。研究结果表明:此法编程时间短,利用该编程求解,解的精度更高,计算效率提高了近100倍,便于引入其他性质,增强标定电子结构的准确性。
关键词:单原子状态自洽法;算法;MATLAB
中图分类号:TG146.4 文献标识码:A 文章编号:1672-7207(2007)05-0900-06
Algorithms of single-atom self-consistency method
PENG Hong-jian1, 2, XIE You-qing2, LI Xiao-bo2
(1. School of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
2. School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: Systematic analysis of algorithms of single-atom self-consistency method shows that traditional algorithms have the following the problems: the accuracy of answer is not high and the time of calculation is too long and so on. The MATLAB (MATrix LABoratory) software aggregated the strongest value calculation as a tool. Take metal Cu for example. The solution is introduced for one-atom self-consistency for determining electronic structure of crystals which finished making program in a short time. The answer is more accurate than ever. Its efficiency is enhanced almost by one hundred times. It is convenient to add other properties to accurately determine the electronic structure of crystal.
Key words: single-atom self-consistency method; algorithm; MATLAB
谢佑卿等经过研究,建立了合金系统科学框架,它由纯金属系统科学,合金物理与化学和合金统计热力学3部分组成,其核心内容是纯金属单原子(OA)理论[1-6]和合金特征晶体理论(CC)[7-10],单原子状态自洽法是在余瑞璜发展的Pauling键长公式[11],吕振家等建立的结合能公式[12-13]的基础上建立的确定晶体电子结构的计算方法[14]。长期以来,研究者用BASIC和FORTRAN等语言编程解决问题,耗费大量的时间用于编程,而有的编程人员不懂专业,这导致编制的程序不是最优秀的,存在解的精度不高和计算时间长等问题,且所选的基本原子态数目不能太多,否则导致求解太慢而影响分析速度。在此,本文作者以MATLAB为工具[15],利用其中集成了最优秀算法的函数来解非线性方程和非线性方程组(只定义方程,输入参数就求解),这样减少了繁琐的编程,而把时间和精力放在模型建立和参数定义的研究上,且由于采用最优秀的算法,解的精度和计算效率提高了许多,很好地满足该方法扩展的需要。
1 单原子状态自洽法的原理
根据纯金属的晶格常数(a)和结合能(Ec)实验值来确定晶体的电子结构,即通过 a和Ec方程来确定键长、键数和键上电子对数[16]。其基本方程为

式中:nc和R(1)是杂化原子的共价电子数和单键半径;rs,Is和ns是各类键的键长、键数和键上共价电子对数;Gs和Is是与晶格类型相关的常数;β按EET理论取值[11]:
 (2)
(2)
关于β的具体取值要通过自洽得到,β值迭代示意图如图1所示,其中flag表示判断β值收敛的语句。
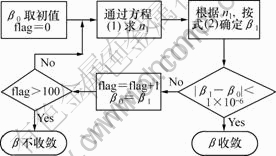
图1 β值迭代示意图
Fig.1 Schematic diagram for overlapping of β value
在平衡状态,rs=rs, 0,1 mol晶体的结合能Ec为:

在纯金属中,每1个原子一般由2个或多个基本原子状态杂化而成。每1个基本原子状态中的外层电子可分为共价电子、近自由电子、磁电子和非键电子。在每1个基本原子态中,电子分布均遵守Pauli不相容原理,电子占有数满足如下的关系:
 (4)
(4)
式中:下标n和l分别为主量子数和角量子数;nT为核外总电子数。
纯金属的杂化原子状态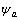 由若干基本原子态
由若干基本原子态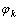 组成:
组成:
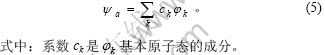
以 和
和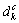 分别表示k基本态中s和d轨道中的共价电子数,
分别表示k基本态中s和d轨道中的共价电子数, 和
和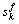 分别表示相应轨道上的非键电子数和近自由电子数,Rk为基本原子态的单键半径,则纯金属的单原子状态参数可由下式求得:
分别表示相应轨道上的非键电子数和近自由电子数,Rk为基本原子态的单键半径,则纯金属的单原子状态参数可由下式求得:
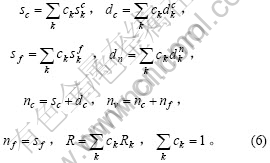
2 单原子状态自洽法的算法
若纯金属由k态杂化组合得到,则问题可归结为解一个非线性方程组,
 (7)
(7)
式中:a为晶格常数的实验值;Ec为结合能的实验值;a(c1, c2, c3)为晶格常数关于c1, c2和c3的函数;E(c1, c2, c3)为结合能关于c1, c2和c3的函数。
当k=2时,双态杂化,方程(6)为超定方程组,无精确解,只有最小二乘解。
当k=3时,三态杂化,方程(6)为洽定方程组,若有解,则为精确解。
当k>3时,多态杂化,方程(6)为欠定方程组,有无穷多解。
因此,采用三态杂化求解来确定晶体的电子 结构。
图2 所示为单原子状态自洽法流程图,其中2个较难的算法需用MATLAB中的函数来解决。

图2 单原子状态自洽法流程图
Fig.2 Schematic procedure for single-atom self-consistency method
第1个是在求基本态中解非线性方程(1),利用fzero函数,函数所采用的算法是二分法、secant法和逆二次内插法的组合。其基本调用形式为
x=fzero(fun, x0, options, P1, P2, …)。
其中:x为函数的解;x0为函数解的初始值;fun为非线性方程的表达式;options为控制参数。变量P1, P2, …等一并传递给目标函数fun。
第2个是在求杂化态中解非线性方程(7),利用fsolve函数,函数所采用的算法是非线性最小二乘法。使用最小二乘法的好处是如果等式系统没有零解,该算法仍然返回一个残差很小的点。其基本调用形式为
x=fsolve(fun, x0, options, P1, P2,…)。
其中:x为函数的解,x0为函数解的初始值(以向量形式表达);fun为非线性方程组的表达式;options为控制参数。变量P1, P2, …等一并传递给目标函数fun。
3 单原子状态自洽法程序的特点
a. 计算的精度提高。
可以通过options参数控制逼近实验值a和Ec的精度,一般为1×10-6。金属Cu的基本原子态及赝晶体的特征性质如表1所示,与以前的计算[17]比较,选同样的基本态,杂化也选同样的三态,而计算结果更准确,且还能找出满足此精度(1×10-6)的其他4个解,与第一原理计算结果相近[18]。
表1 金属Cu的基本原子态及赝晶体的特征性质
Table 1 Basic atomic states and corresponding pseudo crystal characteristic properties of Cu metal
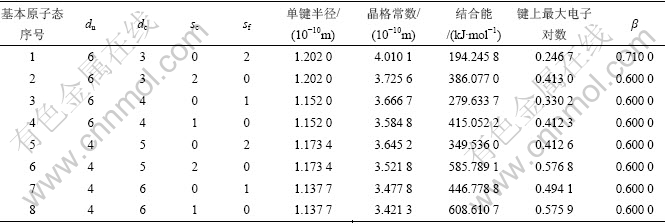
表2 金属Cu的原子状态参数、键参数和特征性质
Table 2 Atomic state parameters, bond parameters and characteristic properties of Cu metal

b. 计算效率提高了近100倍。
若不对基本态中的dn加以限制,则金属Cu的基本态有26个,满足条件的三态杂化解有70个,计算时间约为850.3 s(在CPU为PⅢ750,内存为128 MB计算机上计算的)。如果考虑磁电子或p轨道电子,那么基本态的数目还将增多,计算效率的提高为此提供了计算条件。
c. 可扩展性强。
如果有n种性质来确定晶体电子结构,且要精确满足n种性能的值,一般需要n个基本态杂化才可解决。本解法只需将方程组(7)扩展,将新性质的方程包含进去就可解决。如对于磁性金属,方程组(7)扩展为:
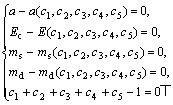 (8)
(8)
由于实验值存在误差,且每个实验值误差精度不一样,精确地满足每一个实验值没有必要,只要大致满足在每一个实验值的精度范围之内即可。可以通过三态杂化来满足多种性质,因为解非线性方程组时采用非线性的最小二乘法也能满足这种要求。将方程组(8)改为方程组(9):
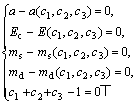 (9)
(9)
这种扩展不是最好的,因为它只控制了一个反映各种性质总的误差,还需要手动从中去挑选满足各种性质精度的解。更好的方法是采用fgoalattain函数替代fsolve函数。fgoalattain函数可以控制每一个目标到达的精度。
由于使用了成熟和优秀的算法,且采用函数调用的形式,使编程从数学问题解脱出来,着重考虑问题的物理模型和参数的选择,如单键半径和屏蔽常数的选择、过渡元素是否考虑p轨道等问题,这些方面只要简单修改程序就可以实现,便于对新问题进行尝试。
4 结 论
a. 利用数值运算功能极强的MATLAB为工具,以金属Cu为例,对确定晶体电子结构的计算方法单原子状态自洽法编程求解,能在短时间内完成编程,解的精度更高,计算效率提高了近100倍,并且能满足该方法扩展的需要。
b. 通过晶格常数和结合能标定电子结构时,存在多解的现象,应增加标定电子结构的准确性。
参考文献:
[1] XIE You-qing. The framework of metallic materials systematic science[J]. Mater Rev, 2001, 15(4): 12-15.
[2] 谢佑卿, 马柳莺. 晶体价电子结构的理论晶格参量[J]. 中南矿冶学院学报, 1985, 16(1): 1-10.
XIE You-qing, MA Liu-ying. The theoretical lattice parameter of valence electron structure of crystal[J]. J Cent South Inst of Mining and Metall, 1985, 16(1): 1-10.
[3] XIE You-qing. A new potential function with many-atom interactions in solid[J]. Science in China: Series A, 1993, 36(1): 90-99.
[4] XIE You-qing. Electronic structure and properties of pure iron[J]. Acta Metall Mater, 1994, 42(11): 3705-3714.
[5] 彭红建, 谢佑卿. 贵金属Ir的原子状态与物理性质[J]. 中南大学学报: 自然科学版, 2006, 37(5): 937-941.
PENG Hong-jian, XIE You-qing. Atomic states and physical properties of rare metal Ir[J]. J Cent South Univ: Science and Technology, 2006, 37(5): 937-941.
[6] 陶辉锦, 谢佑卿, 彭红建. 面心立方和液体Au的热力学性质[J]. 中南大学学报: 自然科学版, 2006, 37(3): 537-542.
TAO Hui-jin, XIE You-qing, PENG Hong-jian, Thermodynamic properties of FCC and Liquid Au[J]. J Cent South Univ: Science and Technology, 2006, 37(3): 537-542.
[7] Xie Y Q, Peng K, Liu X B. Influences of xTi/xAl on atomic states, lattice constants and potential energy planes of ordered FCC TiAl type alloys[J]. Physica B, 2004, 344: 5-20.
[8] Xie Y Q, Liu X B, Peng K. Atomic states, potential energies, volumes, brittleness and phase stability of ordered FCC TiAl3 type alloys[J]. Physica B, 2004, 353: 15-33.
[9] Xie Y Q, Peng H J, Liu X B. Atomic states, potential energies, volumes, brittleness and phase stability of ordered FCC Ti3Al type alloys[J]. Physica B, 2005, 362: 1-17.
[10] Xie Y Q, Tao H J, Peng H J, Atomic states, potential energies, volumes, brittleness and phase stability of ordered FCC TiAl2 type alloys[J]. Physica B, 2005, 366: 17-37.
[11] 余瑞璜. 固体与分子经验电子理论[J]. 科学通报, 1978, 23(4): 217-219.
YU Rui-huang. Solid and molecule experiential electron theory[J]. Chinese Science Bulletin, 1978, 23(4): 217-219.
[12] 吕振家, 王绍铿. 碱金属和碱土金属结合能的计算[J]. 科学通报, 1979, 24(8): 742-745.
L? Zhen-jia, WANG Shao-keng. Calculations on cohesive energies of alkali and alkaline-earth metals[J]. Chinese Science Bulletin, 1979, 24(8): 742-745.
[13] 徐万东, 张瑞林, 余瑞璜. 过渡金属化合物晶体结合能计算[J]. 中国科学: A辑, 1988, 31(3): 323-330.
XU Wan-dong, ZHANG Rui-lin, YU Rui-huang. Calculations on cohesive energies of transitional metal[J]. Science in China: Series A, 1988, 31(3): 323-330.
[14] 谢佑卿, 马柳莺, 张晓东. 确定晶体电子结构的单原子状态自洽法[J]. 科学通报, 1992, 37(16): 1529-1532.
XIE You-qing, MA Liu-ying, ZHANG Xiao-dong. One atom self-consistency method for determining electronic structure of crystal[J]. Chinese Science Bulletin, 1992, 37(16): 1529-1532.
[15] 飞思科技研发中心. MATLAB 6.5辅助优化计算与设计[M]. 北京: 电子工业出版社, 2003: 11-25.
The Center of Research of FECIT Sci-Tec. Auxiliary optimization computation and designations of MATLAB 6.5[M]. Beijing: Publishing House of Electronics Industry, 2003: 11-25.
[16] 谢佑卿. 金属材料系统科学[M]. 长沙: 中南工业大学出版社, 1998: 25-98.
XIE You-qing. Metallic materials systematic science[M]. Changsha: Central South University of Technology Press, 1998: 25-98.
[17] 谢佑卿, 张晓东. 金属Cu 电子结构和物理性质[J]. 中国科学: A辑, 1993, 36(5): 545-552.
XIE You-qing, ZHANG Xiao-dong. Electronic structure and physical properties of Cu metal[J]. Science in China: Series A, 1993, 36(5): 545-552.
[18] Eckardt H, Fritsche L, Noffke J. Self-consistent relativistic band structure of the noble metals[J]. J Phys F: Met Phys, 1984, 14: 97-112.
收稿日期:2006-12-12;修回日期:2007-01-28
基金项目:国家自然科学基金资助项目(50471058);湖南省科技计划资助项目(06FJ3033)
作者简介:彭红建(1972-),男,湖南湘潭人,博士研究生,从事金属材料的研究
通信作者:彭红建,男,博士研究生;电话:0731-8879287;E-mail:phj108@163.com

