中国有色金属学报 2004,(04),534-538 DOI:10.19476/j.ysxb.1004.0609.2004.04.003
低铝含量Ni75 Alx V25-x 合金早期沉淀过程的原子尺度计算机模拟
赵宇宏 陈铮 王永欣 唐丽英
西北工业大学材料科学与工程学院,西北工业大学材料科学与工程学院,西北工业大学材料科学与工程学院,西北工业大学材料科学与工程学院 西安710072 ,西安710072 ,西安710072 ,西安710072
摘 要:
采用微观相场模型,基于离散格点形式的微观扩散方程和非平衡自由能函数,编制了三元Ni75AlxV25-x合金的原子层面计算机模拟程序。模拟发现:低铝含量Ni75AlxV25-x合金的θ相先于γ′相析出,其沉淀机制为等成分有序化+失稳分解;γ′相在θ相的相界处非经典形核,二者均先形成非化学计量比有序相,之后向化学计量比有序相转变。
关键词:
早期沉淀 ;原子尺度 ;计算机模拟 ;
中图分类号: TG111
作者简介: 赵宇宏(1974),女,博士.;
收稿日期: 2003-03-25
基金: 国家自然科学基金资助项目(50071046);
Atomic-scale computer simulation for early precipitation process of Ni75 Alx V75-x alloys with lower Al concentration
Abstract:
With the microscopic phase-field model, the atomic-scale computer simulation programs of the ternary Ni-based alloys were worked out based on the microscopic diffusion equation and nonequilibrium free energy. The results show that for Ni75 Alx V25-x alloy with lower Al composition, θ ordered phase precipitates earlier than γ′ ordered phase does by congruent ordering and spinodal decomposition mechanism, and thus produces a nonstoicheometric θ single ordered phase. Then, the nonstoicheometric γ′ phase precipitates by a non-classical nucleation and growth mechanism at the APBS of θ phases.
Keyword:
early precipitation process; atomic-scale; computer simulation;
Received: 2003-03-25
合金早期沉淀的时间尺度在秒级, 空间尺度在数十至数百原子, 即形成纳米级弥散强化相, 现有的实验手段研究合金沉淀早期的规律和机制非常困难, 采用原子层面的计算机模拟研究具有必要性, 并显示出实验手段无可比拟的优越性。 Pareige
[1 ]
和Poduri
[2 ]
等分别采用蒙特-卡洛法及相场法简单研究了Ni-Al-V合金的沉淀序列, 但均没有对合金沉淀早期的不同机制、 原子尺度的有序-无序转变及非平衡现象作深入探讨。
本文作者基于三元体系微观扩散方程, 通过对沉淀过程中原子图像、 序参数等的模拟和计算分析, 对低铝含量Ni-Al-V体系早期沉淀过程中γ ′相(Ni3 Al)和θ 相(Ni3 V)共存时有序相的析出序列及不同沉淀机制进行了研究。
1三元体系微观相场扩散方程
该模型首先由Khachatuyran
[3 ]
提出, Chen等
[4 ,5 ,6 ,7 ]
做了发展。 设p A (r , t ), p B (r , t ), p C (r , t )分别为A, B和C原子在t 时刻占据格点位置r 的几率, 由于p A (r , t )+p B (r , t )+p C (r , t )=1, 所以每个格点上只有2个方程是独立的。 假设以A和B原子的占位几率为2个独立变量, 微扩散方程为
{
d
p
A
(
r
,
t
)
d
t
=
1
k
B
Τ
∑
r
′
[
L
A
A
(
r
-
r
′
)
?
F
?
p
A
(
r
′
,
t
)
+
L
A
B
(
r
-
r
′
)
?
F
?
p
B
(
r
′
,
t
)
]
d
p
B
(
r
,
t
)
d
t
=
1
k
B
Τ
∑
r
′
[
L
B
A
(
r
-
r
′
)
?
F
?
p
A
(
r
′
,
t
)
+
L
B
B
(
r
-
r
′
)
?
F
?
p
B
(
r
′
,
t
)
]
?
?
?
(
1
)
式中 L αβ r -r ′)为与单位时间内1对α 和β 原子在格点位置r 和r ′上的交换几率有关的常数, α , β =A, B或C; F 为体系的总自由能, 采用平均场自由能; T 为温度; k B 为玻尔兹曼常数。
式(1)为确定方程, 不能描述形核过程, 需添加随机项来模拟热起伏, 使之变为随机方程, 并作傅立叶变换后, 得到傅立叶空间中的微观Langevin方程
{
d
p
?
A
(
k
,
t
)
d
t
=
1
k
B
Τ
∑
r
′
[
L
?
A
A
(
k
)
{
?
F
?
p
A
(
r
′
,
t
)
}
k
+
L
?
A
B
(
k
)
{
?
F
?
p
B
(
r
′
,
t
)
}
k
]
+
ξ
(
k
,
t
)
d
p
?
B
(
k
,
t
)
d
t
=
1
k
B
Τ
∑
r
′
[
L
?
B
A
(
k
)
{
?
F
?
p
A
(
r
′
,
t
)
}
k
+
L
?
B
B
(
k
)
{
?
F
?
p
B
(
r
′
,
t
)
}
k
]
+
ξ
(
k
,
t
)
?
?
?
(
2
)
式中 k 为第一布里渊区定义的倒易格矢;
p
?
A
(
k
,
t
)
?
p
?
B
(
k
,
t
)
?
L
?
A
A
(
k
)
?
L
?
A
B
(
k
)
?
L
?
B
A
(
k
)
?
L
?
B
B
(
k
)
?
ξ
(
k
,
t
)
为晶格位置坐标r 有关函数的傅立叶变换。
用欧拉方法求解方程, 得到不同时刻的原子占位几率, 进而得到原子图像、 序参数分布等。
2模拟结果与分析
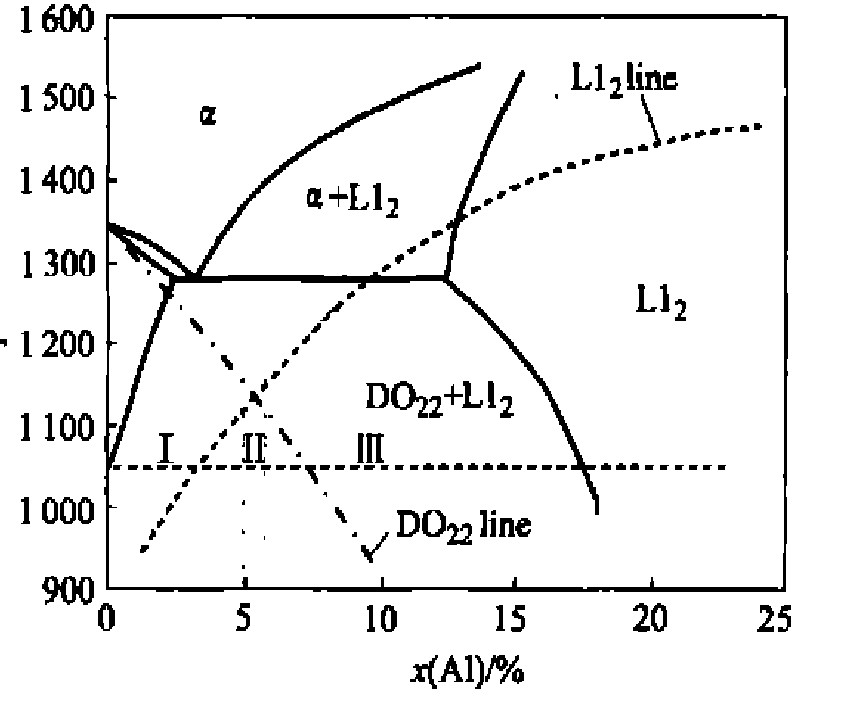
图1所示为由平均场理论计算的Ni3 Al-Ni3 V伪二元相图。 相图上粗实线为平衡相界线, 虚线“L12 line”为L12 相有序化失稳线, 其下方无序相因L12 结构有序化而绝对失稳; 点划线“DO22 line”为DO22 相有序化失稳线, 其下方无序相因DO22 结构有序化而绝对失稳。 2条有序化失稳线交点对应的铝含量为5.3%。 本文将两相场区大致划分为3个区域: Ⅰ区低铝含量合金, 成分范围为0.8%~5.0%; Ⅱ区中间铝含量合金, 成分范围为5.0%~6.0%; Ⅲ区高铝含量合金, 成分范围为6.0%~1.7%。 本文选取位于Ⅰ区的Ni75 Al3.2 V21.8 合金为研究对象。
图1 由平均场理论计算的 Ni-Al-V伪二元Ni3Al-Ni3V相图
Fig.1 Computed pseudobinary Ni3 Al-Ni3 V phase diagram with mean field model
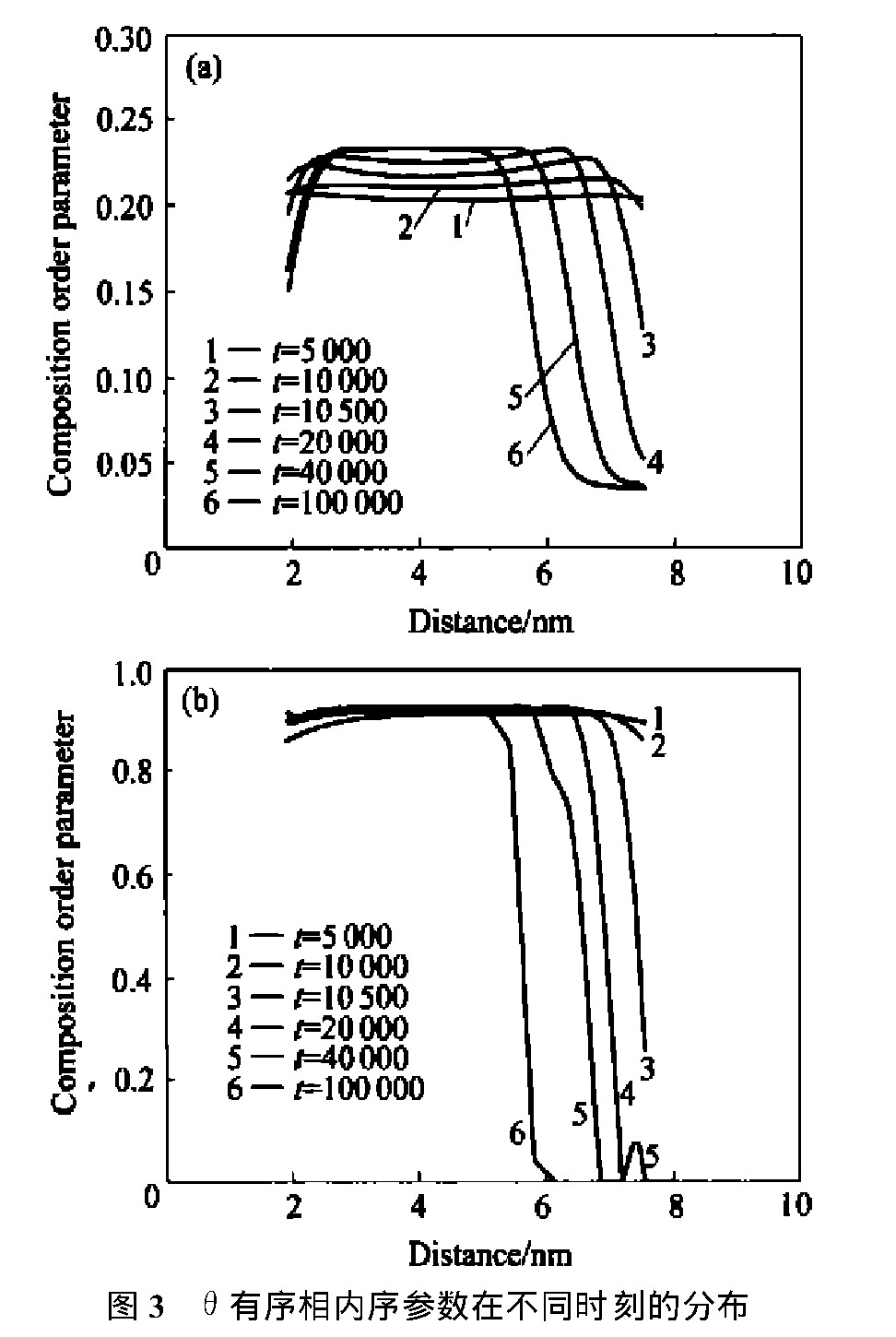
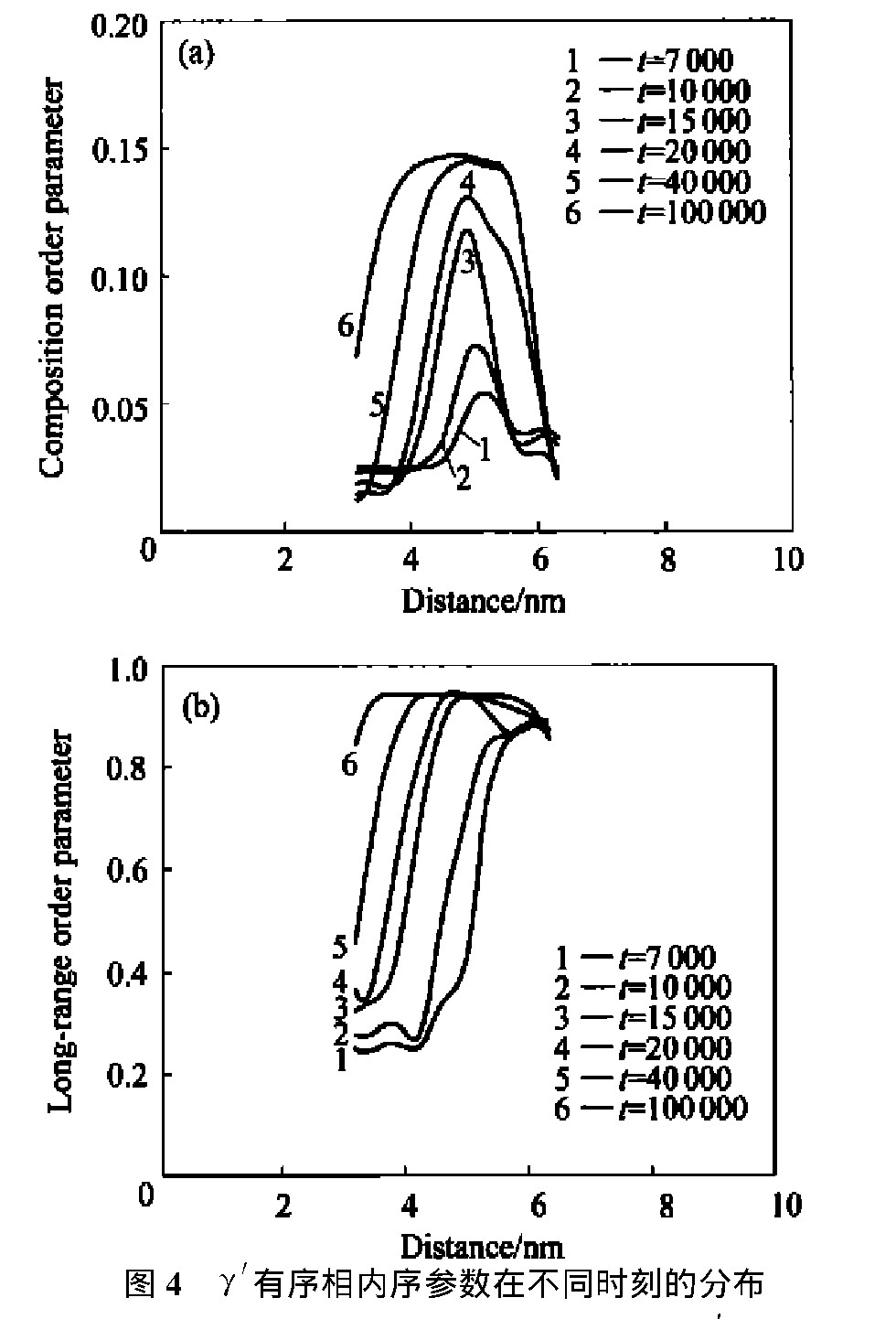
图2所示为不同时间步上该合金的模拟原子图像的演化,图3 和图4所示分别为该合金中θ 相和γ ′相晶核内部的成分序参数和长程序参数分布随时间的变化。
图2所示为铝含量为3.2%时, 不同时间步上合金的模拟原子图像演化过程,时间步长为0.000 2, 格点数为128×128, 镍, 铝和钒原子在每个格点上的占位几率值不同, 从而构成不同的有序结构。 图2(a)的时间步数为2 000, 此时合金仍处于无序基体状态。 图2(b)的时间步数为4 600, 无序基体开始发生有序化转变, 基体中出现一些DO22 有序结构。 图2(c)的时间步数为6 000, 合金中基本完全形成DO22 结构的θ 有序畴及其90°旋转畴, 畴与畴之间被反相畴界(APBS)隔开, 铝原子在θ 相界处聚集。 图2(d)的时间步数为15 000, 在有些APBS处开始出现另一种L12 结构有序畴。 从图2(e)~2(f)中可观察到θ 有序相的进一步粗化、 γ ′相的继续长大及新的γ ′相的形核。
图3中, 在t =5 000时, 成分序参数分布很平, 没有起伏, 而同样时间步数上的长程序参数已经达到最大值, 此为典型的等成分有序化过程, 而且先形成的是非化学计量比θ 有序相, 即成分低于平衡值而长程序参数已达平衡值。 接下来, 成分序参数发生范围甚大而程度较小的起伏, 并逐渐升至平衡值, 变为化学计量比θ 有序相, 且具有明显的中间凹、 两头凸的特征, 是由于所选θ 相左右两边相界处均有γ ′相形核, 钒原子从θ 相界向θ 相内部扩散导致。 随着两边γ ′相的逐渐长大, 此θ 相变小, 反映在序参数变窄, 相内成分序参数分布逐渐变平, 说明钒原子已扩散至θ 相中并达到平衡。
图2 铝含量为3.2%的合金在1 050 K时效时溶质原子占位几率随时间的演化Fig.2 Temporal evolution of occupation probabilities solute atoms at 1 050 K for alloy with 3.2%Al
(a)―t=2 000;(b)―t=4 600;(c)―t=6 000;(d)―t=15 000;(e)―t=20 000;(f)―t=100 000
图3 θ有序相内序参数在不同时刻的分布Fig.3 Order parameter profiles acrossθordered phase for alloy with 3.2%Al at different times
(a)―Composition order parameter;(b)―Long-range order parameter
图4 γ′有序相内序参数在不同时刻的分布Fig.4 Order parameter profiles acrossγ′ordered phase for alloy with 3.2%Al at different times
(a)―Composition order parameter;(b)―Long-range order parameter
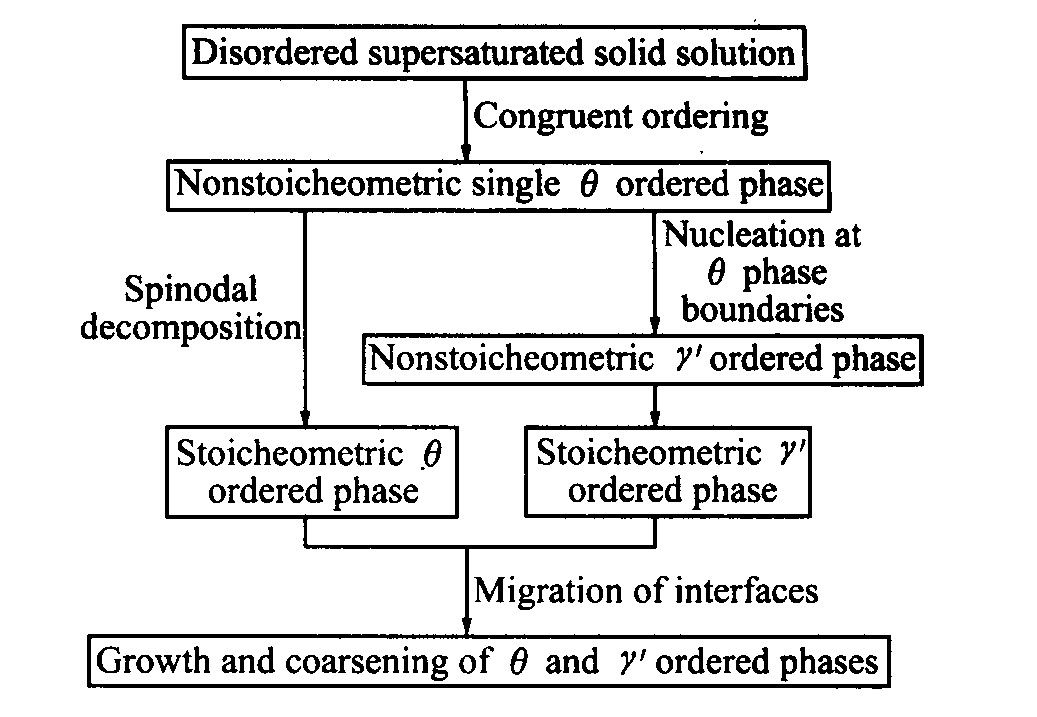
从图4可见, 该γ ′有序相内成分序参数分布满足非经典形核的特点: 核心成分值较低, 远小于平衡值; 成分分布在相界面处有一定的空间延展尺度。 结合长程序参数, 认为先形成非化学计量比γ ′相。 随着时效时间的延长, 核心成分值逐渐接近平衡, 转变为化学计量比γ ′相。 Ni75 Al3.2 V21.8 合金的沉淀过程如图5所示。
3与相似实验的对照
美国宾夕法尼亚州大学Bendersky等
[8 ]
在对Ni75 Alx 25-x 合金体系在1 073 K时效早期的实验观察中, 发现Ni3 Al和Ni3 V共存的组织形态, 认为Ni3 Al和Ni3 V构成伪二元系。 本文的模拟结果与其实验研究一致。
Zapolsky等
[9 ]
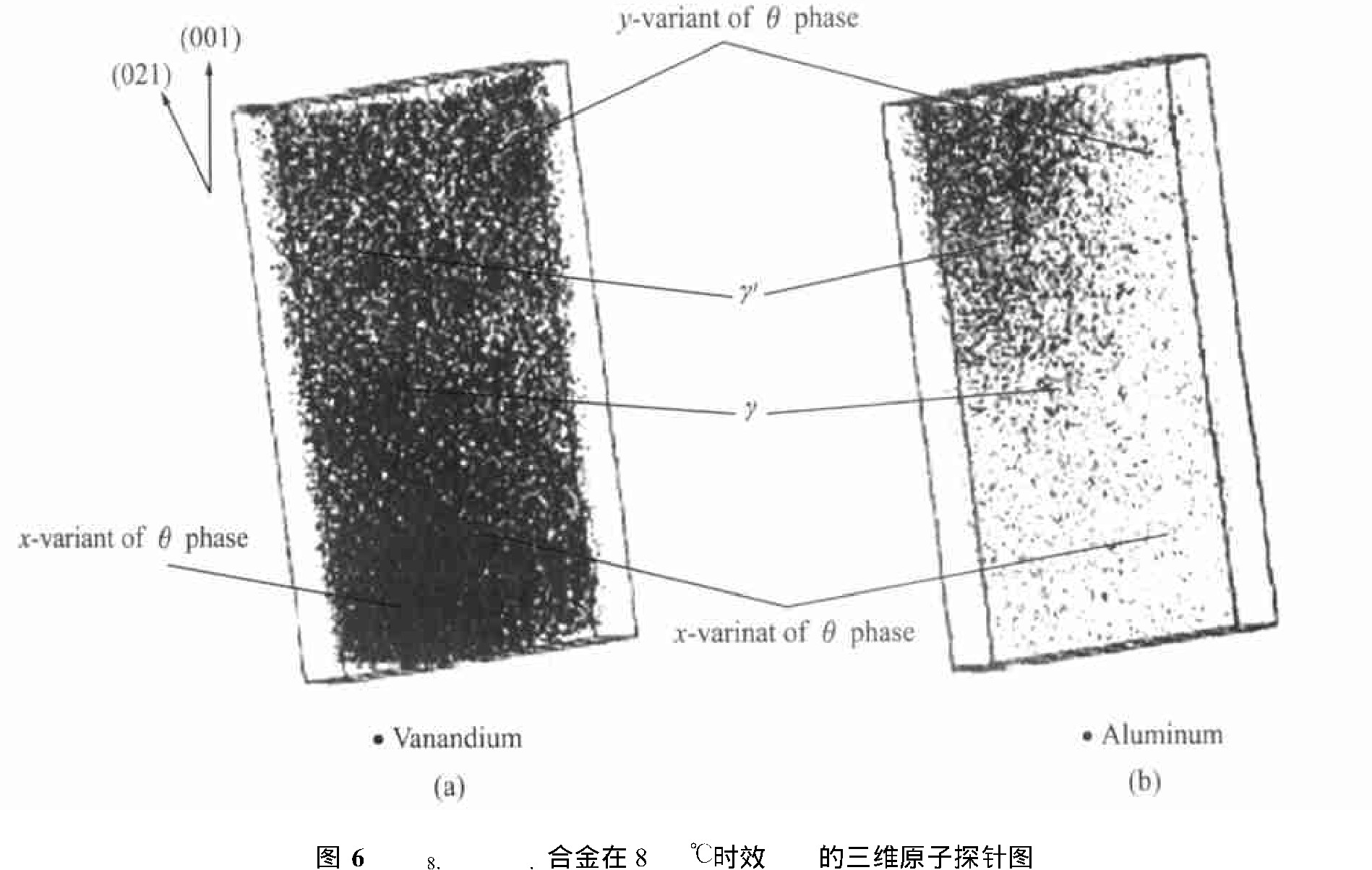
利用三维原子探针分析(3DAP)对三元Ni78.5 Al7 V14.5 体系在800 ℃等温时效时的转变路径进行了研究, 其研究发现, 沉淀过程中DO22 结构的θ 相(Ni3 V)和L12 结构的γ ′相(Ni3 Al)同时析出。 图6所示为该研究提供的2种有序相的三维原子探针图。
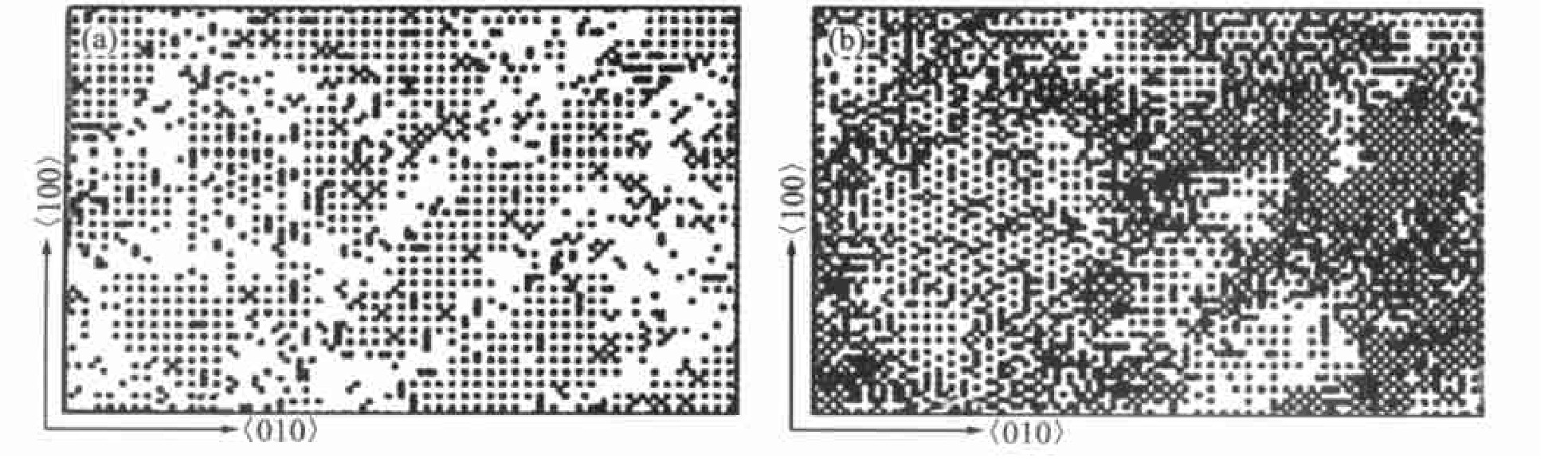
图7所示为Pareige采用蒙特卡洛方法对Ni78.3 Al6.6 V15.1 合金在800 ℃等温时效的早期过程进行研究
[1 ,2 ,6 ]
所得的三维原子图, 同样发现存在FCC→FCC+L12 +DO22 动力学反应, 体系中θ 相(Ni3 V)和γ ′相(Ni3 Al)同时析出。
图5 Ni75Al3.2V21.8合金的沉淀过程
Fig.5 Precipitate process of Ni75 Al3.2 V21.8 alloy
图6 Ni78.5Al7V14.5合金在800℃时效1 h的三维原子探针图Fig.6 3D atomic probe maps for Ni78.5Al7V14.5alloy aged at 800℃for 1 h
(a)―θphase;(b)―γ′phase
图7 Ni78.3Al6.6V15.1合金在800℃时效时的模拟图Fig.7 3D elemen tmaps of Ni78.3Al6.6V15.1alloy aged at 800℃with Monte-Carlo simulation
(a)―L12 long ordered zones;(b)―DO22 long ordered zones
4结论
1) 低铝含量Ni75 Alx 25-x 合金的无序相有序化动力学过程中有2种有序相沉淀析出, 分别是DO22 结构的θ 有序相和L12 结构的γ ′有序相, 2种有序相构成伪二元体系。
2) 2种有序相的析出序列为: θ 有序相先于γ ′有序相析出。
3) 2种有序相的沉淀过程为: 原子的等成分有序化导致形成DO22 结构的单相θ 有序畴, 畴与畴之间被反相畴界(APBS)隔开, 随后发生失稳分解, 铝原子开始在θ 有序相的相上聚集, 同时钒原子从APBS上减少。 随着铝原子在相上聚集程度的增加, γ ′有序相开始形核和长大。 所以θ 相的沉淀机制为等成分有序化+失稳分解, γ ′相的沉淀机制为异相非经典形核。
4) 两者均先形成非化学计量比有序相, 然后向化学计量比有序相转变。
参考文献
[1] PareigeC,BlavetteD.SimulationoftheFCC→FCC+L12+DO22kineticreaction[J].ScriptaMater,2001,44(2):243247.
[2] PoduriR,ChenLQ.Computer simulationofatomicorderingandcompositionalclusteringinthepseudobinaryNi3Al Ni3Vsystem[J].ActaMater,1998,46(5):17191729.
[3] KhachatuyranAG.TheoryofStructuralTransformationsinSolids[M].NewYork:Wiley,1983.139.
[4] MiyazakiT,KoyamaT,KozakaiT.Computersimulationofthephasetransformationinrealalloysystemsbasedonthephasefieldmethod[J].MaterSciEngA,2001,A312:3849.
[5] PoduriR,ChenLQ.Computersimulationofmorphologicalevolutionandcoarseningofδ′(Al3Li)precipitatesinAl Lialloys[J].ActaMater,1998,46(11):39153928.
[6] ChenLQ.Computersimulationofspinodaldecompositioninternarysystems[J].ActaMetallMater,1994,42(10):35033513.
[7] ChenLQ.Acomputersimulationtechniqueforspinodaldecompositionandorderinginternarysystems[J].ScriptaMetallurgicaetMaterialia,1993,29(5):683688.
[8] BenderskyLA.Solid SolidPhaseTransfomations[M].Warrendale,PA:TMS,1994.899902.
[9] ZapolskyH,PareigeC,MarteauL,etal.AtomprobeanalysesandnumericalcalculationofternaryphasediagraminNi Al Vsystem[J].Calphad,2001,25(1):125134.