
First-principles calculation of structural and elastic properties of Pd3-xRhxV alloys
WANG Tao-fen1, 2, CHEN Ping1, DENG Yong-he1, TANG Bi-yu1
1. Key Laboratory of Low Dimensional Materials and Application Technology of Ministry of Education,
Department of Physics, Xiangtan University, Xiangtan 411105, China;
2. Department of Physics, Hunan University of Science and Technology, Xiangtan 411201, China
Received 20 March 2010; accepted 15 July 2010
Abstract: The structural stability, electronic and elastic properties of Pd3-xRhxV alloys with L12 and D022 structures were investigated theoretically by the first-principles calculations. The results reveal that with the increase of Rh content, the unit cell volume of Pd3-xRhxV alloys with L12 and D022 structures decreases, and the structure of Pd3-xRhxV alloys tends to transform from D022 to L12. The elastic parameters such as elastic constants, bulk modulus, shear modulus, elastic modulus, and Poisson ratio, were calculated and discussed in details. Electronic structures were also computed to reveal the underlying mechanism for the stability and elastic properties.
Key words: Pd3-xRhxV alloys; first-principle calculations; electronic structure; elastic properties
1 Introduction
The transition metals and their inter-metallic alloys are promising structural materials because of their high melting points, high strength, and good oxidation resistances[1]. In recent years, the inter-metallic alloys have attracted considerable experimental and theoretical interests of many researchers[2]. Experimental results on many inter-metallic alloys reveal that the L12 atomic order is significantly more ductile than the D022 order, due to the lack of a sufficient number of slip systems in the D022 structure[3]. Consequently, the L12 alloys are more suitable in structural applications because of their excellent mechanical properties[4]. Whereas the D022 structure is an ordered tetragonal phase and closely related to the L12 structure (by a 1/2 [110] shift on every (001) plane). Through part replacement of high e/atom atoms in D022-based alloys with low e/atom atoms, the D022-based alloys can be transformed into the L12 structure[5]. Therefore, it is expected that the mechanical properties of these modified D022-based alloys can be improved because of the availability of more slip systems in the L12 structure[6].
Recent studies proved that it is possible to modify the microstructure of D022. For example, LIU[7] found that although Ni3V compound is prone to crystallize into the tetragonal D022 structure. by adding Co and Fe into Ni3V, the pseudobinary compound (Ni, Co, Fe)3V can be stabilized either in the cubic L12 structure or in the tetragonal D022 structure, depending on the Co or Fe content. The reason is that Co or Fe has lower e/atom than Ni, so the addition of Co or Fe to Ni3V can lower the overall e/atom to an appropriate value. In addition, similar experimental study was performed for Pd3V and Rh3V compounds[8]. It was found that the Pd3V alloys are prone to crystallize into the tetragonal D022 structure, while the Rh3V alloys prefer to L12 structure[8-9]. Hence, it is expected that substituting Pd by Rh can stabilize the L12 structure and further improve the mechanical properties of Pd3V-based compounds.
Nowadays, ab initio calculations are extensively used to predict the structure, stability and mechanical properties of pseudobinary alloys[10]. In order to get a better understanding of structural stability and mechanical properties of Pd3-xRhxV alloys after the substitution of Pd with Rh in Pd3V alloys, ab initio theoretical calculations of Pd3-xRhxV inter-metallic compounds are necessary. The main aim of this study is to investigate the effect of substitution of Pd with Rh on the structural stability and mechanical properties, and provide valuable theoretical results to optimize and design the transition metal alloys.
2 Method of calculation
All calculations were done by using the Vienna ab-initio Simulation Package (VASP) program[11], which is based on density functional theory (DFT). The Perdew�CWang (PW91) version of the generalized gradient approximation (GGA)[12] was used to describe the exchange correlation function, and the projector augmented wave (PAW) method[13] was used in the present work. The cutoff energy (Ecut) of atomic wave functions was set at 350 eV. The Brillouin zone integrations used Monkhorst�CPack grids [14] of 12��12��12 for L12 and 12��12��8 for D022, respectively. The k-point was increased to 18��18��18 (L12) and 18��18��12 (D022) for the density of states (DOS) calculation. Atomic geometry optimization was performed by full relaxation with the conjugate gradient method until the total energy changes within 10-5 eV/atom and the Hellmann-Feynman force on all atomic sites was than 10-2 eV/?. The first-order Methfessel-Paxton[15] method with a width of 0.2 eV was used for total energy calculations, and the calculations of density of states (DOS) were performed with the linear tetrahedron method with Bloch correction[16-17]. The elastic constants were obtained by calculating the total energy as a function of appropriate lattice deformation[18].
3 Results and discussion
3.1 Structural parameters
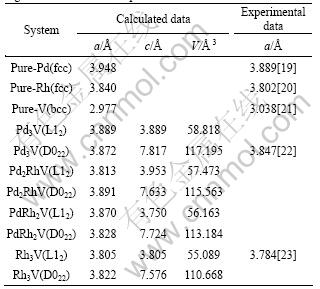
The structural optimization was firstly performed with the conjugate-gradient algorithm by full relaxation of unit cell volume and shape as well as the internal atomic positions. The equilibrium lattice constants were obtained from the minimum total energy. The lattice parameters for pure Pd, Rh, V and Pd3-xRhxV are reported in Table 1, together with the experimental and other theoretical results in Refs.[19-23]. For pure metal Pd, Rh, V, and alloys Pd3V (D022) and Rh3V (L12), the calculated values of lattice constants are in good agreement with the experimental values, implying that the calculation parameters chosen in this work are valid. In addition, it is noticeable that as the Rh content increases, the unit cell volume of Pd3-xRhxV decreases in both the L12 and D022 structures. Therefore, it can be concluded that the substitution of Pd with Rh leads to the contraction of Pd3-xRhxV alloy in both L12 and D022 structures.
Table 1 Calculated lattice constants, equilibrium volume V together with available experimental data
3.2 Formation heat and cohesive energy
For the better understanding of the stability of Pd3-xRhxV alloys, the calculation of formation heat and cohesive energy was performed[24]. The average formation heat of Pd3-xRhxV alloy is defined as[25]:
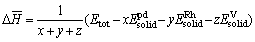 (1)
(1)
where 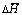 is the average formation heat per atom; Etot is the total energy of the unit cell;
is the average formation heat per atom; Etot is the total energy of the unit cell; 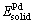 ,
, 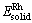 and
and 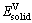 are the energy per atom of bulk Pd, Rh and V, respectively; x, y and z refer to the numbers of Pd, Rh and V atoms in unit cell.
are the energy per atom of bulk Pd, Rh and V, respectively; x, y and z refer to the numbers of Pd, Rh and V atoms in unit cell.
The obtained formation heat of Pd3-xRhxV is shown in Fig.1. From Fig.1, it is found that for L12 structure, the negative formation heat of Pd3-xRhxV becomes lower when x increases from 0 to 3, so the Pd3-xRhxV alloys become more stable from the energetic point of view. While for the D022 structure, with the increase of x, the formation heat of Pd3-xRhxV is also smaller, indicating that the addition of Rh component in binary alloys (Pd3V) also increases the stability of D022 crystal structure. In addition, it should be noted that when x is equal to 0 or 1��the heat formation of D022 structure is more negative than that of L12 structure, so the Pd3V and Pd2RhV are prone to D022 structure. When x is 2 or 3, the heat
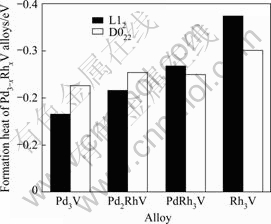
Fig.1 Formation heat of Pd3-xRhxV alloys
formation of L12 structure is more negative than that of D022, hence, the PdRh2V and Rh3V show the preference to L12 structure. Therefore, with the increase of x, the crystal structure of Pd3-xRhxV possesses the transformation tendency from the D022 to L12 structure, which can improve the mechanic properties of the Pd3-xRhxV.
The stability of crystal structure is also correlated to its cohesive energy[26], and the cohesive energy is often defined as energy needed when crystal is decomposed into the single atom. Generally, the lower the cohesive energy is, the more stable the crystal structure is[27]. In this work, the average cohesive energy is calculated with the following formula[28]:
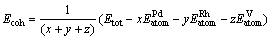 (2)
(2)
where Etot is the electronic total energy of Pd3-xRhxV unit cell; x, y and z are the numbers of Pd, Rh and V atoms in unit cell of Pd3-xRhxV alloy, respectively;  ,
,  and
and are the electronic total energies of single Pd, Rh and V atom in freedom state.
are the electronic total energies of single Pd, Rh and V atom in freedom state.
The obtained cohesive energy of Pd3-xRhxV is listed in Table 2. From Table 2, it is found that for L12 structure, the cohesive energy of Pd3-xRhxV becomes lower when x changes from 0 to 3, so the L12 Pd3-xRhxV would be more stable. For D022 structure, with the increase of x, the cohesive energy of Pd3-xRhxV exhibits a similar variation tendency with that of the L12. So, Pd3-xRhxV is more stable through the substitution of Pd with Rh. From the present results, it is clear that the Pd3V and Pd2RhV are prone to D022 structure, and the PdRh2V and Rh3V show the preference to L12 structure. The results are consistent with the formation heat analyses of Pd3-xRhxV alloys.
Hence, from the results of formation heats and cohesive energies above, it could be expected that with the increase of x, Pd3-xRhxV alloys possess the transformation tendency from D022 to L12 structure by substitution of Pd with Rh in binary alloys (Pd3V), and can further improve the mechanical properties of Pd3V compound. The reasons may be attributed to the fact that the addition of Rh with lower e/atom than Pd can lower the overall e/atom of alloys to an appropriate value.
Table 2 Cohesive energy of Pd3-xRhxV alloys

3.3 Density of states (DOS)
To further reveal the underlying mechanism of the structural stability for the Pd3-xRhxV alloy, the electronic total and partial densities of states (DOS) of alloys were calculated, and the results for the L12 and D022 structures are shown in Fig.2 and Fig.3, respectively.
For comparison and analysis, the total and partial densities of states (DOS) of Pd, Rh and V were also calculated. From Fig.2 and Fig.3, it is seen that for both the L12 and D022 structures of Pd3-xRhxV, the DOS is mainly dominated by d electrons of Pd, Rh and V, and the partial densities of states of Pd (s), Pd(p), Rh(s), Rh(p) and V(s), V(p) in Pd3-xRhxV alloys are much less than the d partial densities of states. Concretely speaking, from the total DOS in Fig.2(a) and Fig.3(a), it is shown that the main bonding peaks of Pd3V for L12 and D022 structure in the energy range under Fermi level (about from -5.4 eV to -0.4 eV) are mainly dominated by valence electron of Pd(d) orbits, and the V(d) valence electron orbits mainly dominates the energy range (about from 0 to 0.8 eV) upon the Fermi level. By further comparison between Figs.2(b) and (c) or Fig.3(b) and (c), it is revealed that the energy range under Fermi level for both the L12 and D022 structures of Pd3-xRhxV (x=1, 2) is mainly affected by the valence electron of both Pd(d) and Rh(d) orbits. Moreover, with the increases of x, the effect of Rh (d) orbits is more prominent under Fermi level. In addition, the electron in low-energy region of Fermi level is related to the stability of crystal structure[14]. Hence, when x increases, the hybridization between Pd-, Rh- and V-d states under Fermi level becomes strong, and the stability of Pd3-xRhxV alloys of both L12 and D022 structure increases.
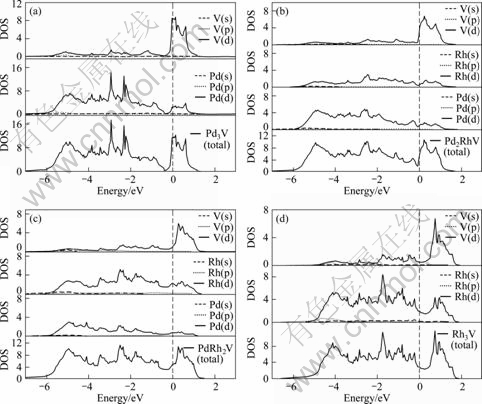
Fig.2 Partial and total densities of states of L12 structure in Pd3-xRhxV alloys (Fermi level is set at zero energy and marked by vertical dashed lines)
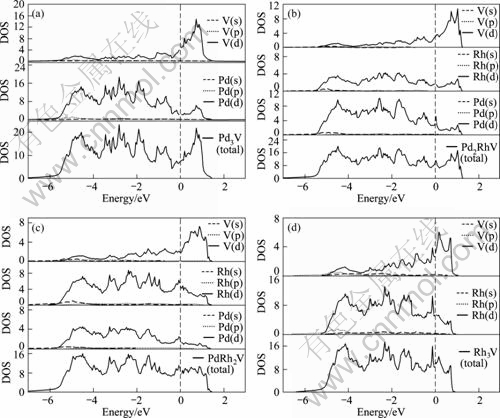
Fig.3 Partial and total densities of states of D022 structure in Pd3-xRhxV (Fermi level is set at zero energy and marked by vertical dashed lines)
3.4 Elastic constants
Generally, the elastic constants are closely related to the mechanical behavior of the materials and also provide the information about the bonding characteristic. The crystal elastic constants can be obtained by the first-principles calculation of the total energy as a function of appropriate lattice deformation. For small strains, Hooke��s law is valid and the crystal energy E is a quadratic function of strain. Thus, to obtain the total minimum energy for calculating the elastic constants to the second order, a crystal is strained and all the internal parameters are relaxed[29].
The calculated values of elastic constants Cij are listed in Table 3. It can be seen from Table 3 that for the Pd3-xRhxV alloys with the L12 structure, all Cij values become larger as x increases. While for D022 structure, the C12 and C66 increase as x increases, implying that the Rh component shows much more effects on the two directions. In addition, for the Pd3-xRhxV alloys in the L12 and D022 structures, the elastic constant C33 is larger than C11, showing that the incompressibility along ��001? is larger than that along ��100?, and the atomic bonding along ��001? is also stronger than that along ��100?.
The requirement of mechanical stability for cubic crystals leads to the following restrictions on the elastic constants[30]:
C11-C12>0, C11+2C12>0, C11>0, C44>0 (3)
And the mechanical stability criteria for tetragonal crystals are[29]
C11-C12>0, C11+C33-2C13>0, 2C11+C33+2C12+4C13 >0,C11>0, C33>0, C44>0, C66>0 (4)
The calculated elastic constants of Pd3-xRhxV alloy in L12 and D022 structures are in good agreement with the above criteria, indicating that all of them are mechanically stable.
Based on the single-crystal elastic constants, the polycrystalline elastic modulus is also estimated by the Voigt-Reuss-Hill (VRH) approximation[31]. For the cubic structure[32], the bulk modulus K, shear modulus G, elastic modulus E, and Poisson ratio (��) are calculated as follows:
K=(C11+2C12)/3 (5)
GV=(C11-C12+3C44)/5 (6)
GR= 5(C11-C12)C44/[4C44+3(C11-C12)] (7)
G=(GV+GR)/2 (8)
E=9GK/(3K+G) (9)
v=(E-2G)/2G (10)
For the tetragonal lattice, the equations are[31]
K=[(C11+C12)C33-2C132]/(C11+C12+2C33-4C13) (11)
GV=(2C11+C33-C12-2C13+6C44+3C66)/15 (12)
GR=15/(8s11+4s33-4s12-8s13+6s44+3s66) (13)
where sij are the compliance constants.
s11+s12=C33/C, s11-s12=1/(C11-C12), s13=-C13/C, s33=(C11+C12)/C, s44=1/C44, s66=1/C66; G=(GV+ GR)/2; E= 9GK/ (3K+G); v= (E-2G)/2G; C=C33(C11+C12)-2C132 (14)
The calculated polycrystalline elastic modulus are listed in Table 4. In general, the elastic modulus can effectively describe the elastic properties of Pd3-xRhxV alloys. The bulk modulus K is a measure of resistance to volume change by applied pressure and the shear modulus G is a measure of resistance to reversible deformations upon shear stress[33]. The larger the K and G are, the higher the resistances to volume change and reversible shear deformations are. The elastic modulus E is used to provide a measure of stiffness of the solid. The larger the elastic modulus is, the stiffer the material is[34]. Table 4 shows that with increasing x, the calculated bulk modulus K, shear modulus G and elastic
Table 3 Crystal elastic constants for L12 and D022 in compounds
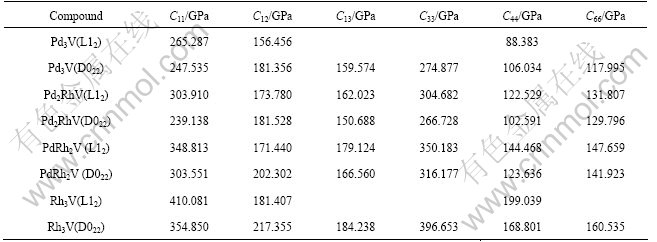
Table 4 Constants of Pd3-xRhxV
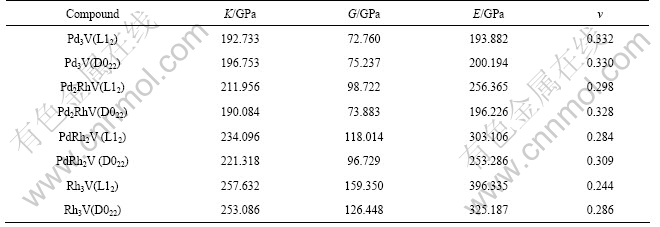
modulus E of L12 Pd3-xRhxV alloys increase. On the other hand, the Poisson ratio is used to quantify the stability of the crystal against shear, which usually ranges from -1 to 0.5. All the calculated Poisson ratios of Pd3-xRhxV (L12) alloys do not exceed 0.35, which means that with increasing x, Pd3-xRhxV alloys still possess good stability against shear. Obviously, the overall elastic properties of Pd3-xRhxV (L12) alloys have been improved with increasing x, which is consistent with the results of LIU[7]. In contrary, for the Pd3-xRhxV (D022) alloys, the elastic modulus does not increase monotonously with increasing x. As seen in Table 4, the elastic modulus of the Pd2RhV (D022) is lower than that of Pd3V, while the elastic modulus of the PdRh2V (D022) is higher than that of Pd3V. In addition, it can be easily found that K, G and E of Pd3-xRhxV (L12) alloys are superior to those of the D022-based alloys with the same x, indicating that the substitution of Pd with Rh is an effective way to improve the elastic properties. Up to now, there is still no report on experimental values of these parameters, so the above theoretical results obtained here provide a reference for the future experimental work.
4 Conclusions
1) The structural stability and elastic properties of Pd3-xRhxV alloys in L12 and D022 structures were investigated based on density-functional theory (DFT) with the generalized gradient approximation (GGA).
2) The results show that the substitution of Pd with Rh leads to contraction of the lattice of Pd3-xRhxV alloys. Furthermore, with the increase of x, L12 crystal structure becomes more stable and the Pd3-xRhxV tends to transform from the D022 to L12 structure, although the substitution of Pd with Rh in binary alloys Pd3V can result in the stable formation of Pd3-xRhxV alloys of both L12 and D022 crystal structure.
3) The calculated electronic structure shows that the hybridization between Rh-d states and V-d states can stabilize L12- and D022-Pd3-xRhxV. In addition, the present investigations show that the elastic properties of L12-based Pd3-xRhxV alloys are improved with the increase of x. In contrary, for Pd3-xRhxV alloys with the D022 structure, the elastic modulus of Pd2RhV decreases, while elastic parameters for PdRh2V increase.
References
[1] COLINET C, PASTUREL A. Phase stability and electronic structure of the HfAl3 compound [J]. Phys Rev B, 2001, 64: 205102.
[2] LEBACQ O, PASTUREL A, MANH D N, FINEL A, CAUDRON R. Electronic structure, cohesive properties and phase stability in Ni3V, Pd3V, and Pt3V compounds [J]. J Alloys Compd, 1998, 264: 31-37.
[3] PETTIFOR D G, COTTRELL A H. Electron theory in alloy design [M]. London: The Institute of Materials, 1992: 158.
[4] SINHA A K. Close-packed ordered ABs structures in ternary alloys of certain transition metals [J]. Trans AIME, 1969, 245: 911-917.
[5] TIAN W H, NEMOTO M. Morphology of L10-TiAl(Ag) precipitation in Ag-modified L12-Al3Ti [J]. Intermetallics, 2000, 8: 345-352.
[6] MORRIS D G, MORRIS M A. The stress anomaly in FeAl-Fe3Al alloys[J]. Intermetallics, 2005, 13: 1269-1274.
[7] LIU C T. Physical metallurgy and mechanical properties of ductile ordered alloys (Fe, Co, Ni) V [J]. Intl Met Rev, 1984, 29: 168-194.
[8] LOISEAU A, CABET E. L12D022 competition in the pseudo binary (Pt, Rh)3V and (Pd, Rh)3V alloys[J]. J Physique IV, 1993, 3: C7-2051.
[9] CABET E, PASTUREL A, DUCASTELLE F, LOISEAU A. L12- DO22 competition in the pseudobinary (Pt, Rh)3V, Pt3(V, Ti), and (Pd, Rh)3V alloys: Phase stability and electronic structure[J]. Phys Rev Lett, 1996, 76: 3140-3143.
[10] COLINET C, PASTURE A. Theoretical calculation of the phase diagram between one-dimensional long-period structures in the quasi binary sections: Pd3xRh3(1-x)V, Pt3xRh3(1-x)V, and Pt3VxTi(1-x) [J]. Calphad, 2002, 26: 563-571.
[11] KRESSE G, FURETHM?LLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Phys Rev B, 1996, 54: 11169-11186.
[12] PERDEW J P, CHEVARY J A, VOSKO S H. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation [J]. Phys Rev B, 1992, 46: 6671-6687.
[13] BLOCHL P E. Projector augmented-wave method [J]. Phys Rev B, 1994, 50: 17953-17979.
[14] DENG Yong-he, WANGT Tao-fen, ZHANG Wei-bing, TANG Bi-yu, ZHNG Xiao-qin, DING Wen-jiang. Crystal structure investigation of Mg3Pd from first-principles calculations[J]. Transactions of Nonferrous Metals Society of China, 2008, 18(2): 41-45.
[15] METHFESSEL M, PAXTON A T. High-precision sampling for Brillouin-zone integration in metals [J]. Phys Rev B, 1989, 40: 3616-3621.
[16] BLOCHL P E, JEPSEN O, ANDERSEN O K. Improved tetrahedron method for Brillouin-zone integrations [J]. Phys Rev B, 1994, 49: 16223-16233.
[17] TANG B Y, WANG N, YU W Y. Theoretical investigation of typical fcc precipitates in Mg-based alloys [J]. Acta Materialia, 2008, 56: 3353-3357.
[18] WALLACE D C, Thermodynamics of crystals [M]. New York: Wiley, 1972: 106-120.
[19] LAWSON A C, CONANT J W, ROBERTSON R. Debye-Waller factors of PdDx materials by neutron powder diffraction[J]. J Alloys Compd, 1992,183: 174-180.
[20] ELLNER M, KOLATSCHEK K, PREDEL B.On the partial atomic volume and the partial molar enthalpy of aluminium in some phases with Cu and Cu3Au structures [J]. Journal of the Less-Common Metals, 1991, 170: 171-184.
[21] KORLING M, HAGLUNG J. Cohesive and electronic properties of transition metals: The generalized gradient approximation[J]. Phys Rev B, 1992, 45: 13293-13297.
[22] DWIGHT A E, DOWNEY J W, CONNER R A. Some AB3 compounds of the transition metals [J]. Acta Crystallographica, 1961, 14: 75-76.
[23] WATERSTRAT R M, MANUSZEWSKI R C. The vanadium-rhodium constitution diagram [J]. Journal of the Less-Common Metals, 1977, 52: 293-305.
[24] SONG Y, GUO Z X, YANG R. Influence of selected alloying elements on the stability of magnesium dihydride for hydrogen storage applications: A first-principles investigation[J]. Phys Rev B, 2004, 69(9): 094205.
[25] ZUBOV V I, SANCHEZ J F, TEIXEIRA J N. Theoretical study of the saturated vapor pressure and enthalpy of sublimation of C60 sfullerite [J]. Phys Lett B, 1997, 55: 6747-6749.
[26] MASSARD R, UZIO D, THOMAZEAU C. Strained Pd overlayers on Ni nanoparticles supported on alumina and catalytic activity for buta-1, 3-dience selective hydrogenation [J]. J Catalysis, 2007, 245: 133-143.
[27] GHOSH G. First-principles calculations of structural energetics of Cu-TM(TM=Ti, Zr, Hf) intermetallics [J]. Acta Materialia, 2007, 55: 3347-3374.
[28] ZUBOV V I, TRETIAKOV N P, TEIXEIRA J N, SANCHEZ J F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a van der Waals crystal��fullerene C60 [J]. Phys Lett A, 1994, 194: 223-227.
[29] PATIL S K R, KHARE S V, TUTTLE B R, BORDING J K, KODAMBAKA S. Mechanical stability of possible structures of PtN investigated using first-principles calculations [J]. Phys Rev B, 2006, 73: 104118.
[30] MATTESINI M, AHUJA R, JOHANSSON B. Cibic Hf3N4 and Zr3N4: A class of hard materials[J]. Phys Rev B, 2003, 68: 184108.
[31] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proc Phys Soc A, 1952, 65: 349-354.
[32] MEHL M J, OSBURN J E, PAPACONSTANTOPOULOS D A, KLEIN B M. Structural properties of ordered high-melting -temperature intermetallic alloys from first-principles total-energy calculations [J]. Phys Rev B, 1990, 41: 10311-10323.
[33] YOUNG A F, SANLOUP C, GREGORYANZ E, SCANDOLO S, HEMLEY R J, MAO H K. Synthesis of novel transitions metal nitrides IrN2 and OsN2 [J]. Phys Rev Lett, 2006, 96: 155501.
[34] MATTESINI M, SOLER J M, YNDURAIN F. Ab initio study of metal-organic framework-5 Zn4O(1, 4-benzenedicarboxylate)3: An assessment of mechanical and spectroscopic properties [J]. Phys Rev B, 2003, 73: 094111.
Pd3-xRhxV�Ͻ�Ľṹ�͵������ܵĵ�һ��ԭ������
���ҷ�1, 2, �� ƽ1, ������1, �����1
1. ��̶��ѧ ����ϵ, ��ά���ϼ���Ӧ�ü����������ص�ʵ����, ��̶411105;
2. ���ϿƼ���ѧ ����ϵ, ��̶411201
ժ Ҫ: ���õ�һ��ԭ����Pd3-xRhxV�Ͻ��2�ֽṹ(L12��D022)������ȶ��ԡ����������Լ��������ܵȷ�������о���������������������������ӣ�L12��D022�ͽṹ�ĺϽ��������С��������һ���̶ȵ�ѹ������Pd3V��ȣ�������Ԫ�أ�������Pd3-xRhxV�Ͻ��2�ֽṹ���ȶ������ҺϽ�ṹ���ڴ�D022��ת��Ϊ�����ȶ���L12�͡��ԺϽ���ӽṹ(̬�ܶ�)�ļ���ͷ���˵����������ļ��룬�ڷ����ܼ�������ͷ����ӻ�����Խ��Խ���ԣ���һ��Ӱ��L12��D022�ṹ���ȶ��ԡ�����Pd3-xRhxV�Ͻ��L12��D022�ṹ�ĵ��Գ����������ģ��(B)������ģ��(G)������ģ��(E)�Ͳ��ɱ�(��)�����������ı仯���ɽ��м�������ۡ�
�ؼ��ʣ�Pd3-xRhxV�Ͻ�; ��һ��ԭ������; ���ӽṹ; ��������
(Edited by YANG Hua)
Foundation item: Project (50861002) supported by the National Natural Science Foundation of China; Project (0991051) supported by the Natural Science Foundation of Guangxi Province, China; Project (08JJ6001) supported by the Natural Science Foundation of Hunan Province, China; Project (KF0803) supported by Key Laboratory of Materials Design and Preparation Technology of Hunan Province, China; Project (X071117) supported by the Scientific Research Foundation of Guangxi University, China
Corresponding author: TANG Bi-yu; Tel: +86-13187321190; E-mail: tangbiyu@xtu.edu.cn
DOI: 10.1016/S1003-6326(11)60726-7

