不同维度草酸亚铁的合成及其组织结构
苏玉长1, 2,陈宏艳1,胡泽星1,刘赛男1,肖立华1,江敏1
(1. 中南大学 材料科学与工程学院,湖南 长沙,410083;
2. 中南大学 有色金属材料科学与工程教育部重点实验室,湖南 长沙,410083)
摘要:采用H2CO4・2H2O和FeSO4・7H2O为原料,通过控制合成条件,合成粒径较小、维度和结构不同的草酸亚铁粉末。采用X线衍射、红外光谱、激光粒度仪和扫描电镜研究陈化温度、加料方式、pH、分散剂用量、搅拌速度、硫酸亚铁浓度等合成条件对产品组织结构、形貌和平均粒径的影响。研究结果表明:室温陈化时,得到纯相β-草酸亚铁,随陈化温度升高,草酸亚铁结构逐渐向α相转变,至90 ℃陈化时,得到纯相α-草酸亚铁;随pH升高,合成草酸亚铁的结晶性变差,但粒径变小;采用硫酸亚铁溶液往草酸溶液里面滴加的进料方式,增加硫酸亚铁浓度、搅拌速度和分散剂与水的体积比,均有利于降低草酸亚铁粉末的平均粒径;通过工艺控制生长,可以合成0维超细粉状、1维杆状和2维片状的草酸亚铁。
关键字:草酸亚铁;维度;组织结构
中图分类号:O611.65 文献标志码:A 文章编号:1672-7207(2013)06-2237-07
Synthesis and microstructure of ferrous oxalate dehydrate of different dimensionalities
SU Yuchang1, 2, CHEN Hongyan1, HU Zexing1, LIU Sainan1, XIAO Lihua1, JIANG Min1
(1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Non-ferrous Materials Science and Engineering of Ministry of Education,Central South University, Changsha 410083, China)
Abstract: Ferrous oxalate dehydrate powders of fine particle size, different microstructures and dimensionalities were prepared using H2CO4・2H2O and FeSO4・7H2O as starting materials. The effects of aging temperature, way of material feeding, pH value, dispersant amount, stirring speed and FeSO4 content on microstructure, particle size and morphology of ferrous oxalate dehydrate prepared were studied by XRD, IR-spectroscopy, laser particle size analyzer and SEM. The results show that pure β-ferrous oxalate dehydrate can be obtained when it is aged at room temperature. As aging temperature increases, products tend to transform to α-ferrous oxalate dehydrate, and pure α-ferrous oxalate dehydrate is obtained when aged at 90 ℃. Crystallinity of ferrous oxalate tends to be worse and particle size gets finer as pH value increases. Adding FeSO4 solution into H2CO4 solution and the increase of FeSO4 content, stirring speed and dispersant amount all generate finer particle size. Crystalline growth can be controlled by different process conditions, and ferrous oxalate dehydrate of zero-dimensional ultra-fine crystalline, one-dimensional rod and two-dimensional flake can be obtained.
Key words: ferrous oxalate dehydrate; dimensionality; microstructure
草酸亚铁是纳米磁性材料、超级电容器用多孔材料及锂电池用磷酸亚铁锂正极材料所需的主要原材料。具有橄榄石结构的磷酸亚铁锂作为新一代锂离子电池正极材料,具有环境友好、原材料来源丰富、成本低、比容量高、充放电过程中结构稳定等优点,是极有前途的锂离子电池正极材料之一[1-2]。但是,磷酸亚铁锂的离子导电率和锂离子扩散系数低、振实密度低、倍率放电性能差、大电流充/放电性能不好等缺点限制其商业应用[3-4]。为克服这些不足,国内外已研究多种合成方法和合成因素[5-7]对磷酸亚铁锂微结构和电性能的影响,其中也包括不同铁源合成磷酸亚铁锂的影响[8]。草酸亚铁作为合成磷酸亚铁锂最常用的铁源之一,具有以下优点:(1) 草酸盐在正极材料合成过程中不易引入杂质相;(2) 草酸亚铁合成的磷酸亚铁锂正极材料结晶度较高,键合力大,有利于稳定样品的骨架结构[9];(3) 草酸亚铁在反应过程中分解产生气体,可阻碍晶粒的长大和团聚[10]。目前,0维粉状、1维纤维或杆状及2维片状等不同维度磷酸亚铁锂的制备及其与电化学性能的关系已成为锂电池用磷酸亚铁锂正极材料的热点课题,草酸亚铁的结构、形貌等对合成磷酸亚铁锂的影响也正处在研究中。商用草酸亚铁均在十几微米,而且尚未见关于草酸亚铁的粒度、形貌和结构等方面的研究和报道。本文作者采用草酸和硫酸亚铁为原料,合成粒径较小、形状各异的草酸亚铁,研究合成过程中的各种反应条件对合成的草酸亚铁的结构、形貌和粒度的影响,以满足锂电池行业对不同维度、结构的草酸亚铁的需求。
1 实验
1.1 实验方法
实验原材料为H2CO4・2H2O(纯度为99.5%) 和FeSO4・7H2O (纯度为99%)。按摩尔比1:1.1称取一定量的FeSO4・7H2O和H2CO4・7H2O,分别用一定量去离子水溶解,制成纯净溶液备用。将一定量的分散剂加入草酸溶液中,混合均匀,在搅拌条件下,将氨水滴入混合溶液,控制pH在1~6之间,搅拌均匀后制成母液。室温下,将硫酸亚铁溶液加入草酸母液中,在此过程中保持搅拌条件。进料完全后,继续搅拌1 h,以使物料充分反应。反应完全后,将产物在不同温度下陈化7 h,后用蒸馏水和无水乙醇进行多次洗涤,固液分离后,于40 ℃干燥24 h,即得到淡黄色草酸亚铁粉末。
1.2 样品的物性表征
采用X线衍射(XRD)、红外光谱(FT-IR)、扫描电镜(SEM)对产物进行组成、结构和形貌表征。X线衍射采用日本理学RigakuDmax-2500VB X线衍射分析仪,以Cu靶的Kα为辐射源(λ=0.154 06 nm),采用步进扫描,步宽为0.02°,扫描范围为10°~80°,扫描速度为8 (°)/min;红外光谱测试使用美国Thermo Scientific 傅里叶变换红外测试仪NICOLET 6700,测量的波数范围为400~4 000 cm-1;采用FEI Sirion200场发射扫描电子显微镜观察产物的微观形貌;采用POP-6型激光粒度仪进行粒度分析。
2 结果与讨论
2.1 草酸亚铁的结构分析
草酸亚铁有2种不同的结构:α-草酸亚铁和β-草酸亚铁。α-草酸亚铁属于单斜晶系,空间群为C2/c(No.15),晶胞参数为a=0.992 1 nm,b=0.555 6 nm,c=0.970 7 nm,β=104.5°;β-草酸亚铁属于正交晶系,空间群为Cccm(No.66),晶胞参数为a=1.226 nm,b=0.557 nm,c=1.548 nm。
图1所示为不同温度下陈化7 h后合成草酸亚铁粉末的XRD图谱。从图1可见:当陈化温度为室温时,所有的衍射峰均可标定为β-草酸亚铁的特征峰(PDF 22-0635),未检索到其他杂质峰。经X线衍射花样指数化和结构精化,其晶胞参数为a=1.222 nm,b=0.555 6 nm,c=1.544 nm。40 ℃和60 ℃陈化得到的草酸亚铁XRD衍射峰与室温陈化样品的衍射峰基本一致,说明低于60 ℃下陈化合成的样品仍为β-草酸亚铁。不同之处在于;随着陈化温度的升高,(202)和(400)晶面的衍射峰位均有向右移动的趋势,而(004)晶面的衍射峰位逐渐向左移动。当陈化温度为80 ℃时,α-草酸亚铁的(002),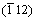 ,
,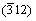 和
和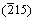 晶面的特征衍射峰开始出现,图1中的衍射峰为α-草酸亚铁和β-草酸亚铁衍射峰的混合。当陈化温度为90℃时,所有的衍射峰均可标定为α-草酸亚铁的特征峰(PDF 23-0293),经X线衍射花样指数化和结构精化,其晶胞参数为a=0.991 5 nm,b=0.555 2 nm,c=0.970 2 nm,β=104.502°。未检测到β-草酸亚铁特征峰和其他杂质峰。
晶面的特征衍射峰开始出现,图1中的衍射峰为α-草酸亚铁和β-草酸亚铁衍射峰的混合。当陈化温度为90℃时,所有的衍射峰均可标定为α-草酸亚铁的特征峰(PDF 23-0293),经X线衍射花样指数化和结构精化,其晶胞参数为a=0.991 5 nm,b=0.555 2 nm,c=0.970 2 nm,β=104.502°。未检测到β-草酸亚铁特征峰和其他杂质峰。
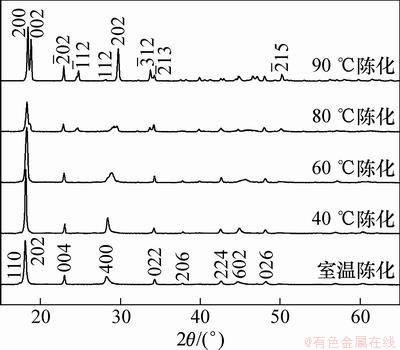
图1 不同温度下陈化7 h的草酸亚铁XRD图谱
Fig. 1 XRD patterns of FeC2O4・2H2O prepared at different aging temperatures for 7 h
上述分析表明:β-草酸亚铁属于热力学上的亚稳相,当温度大于60 ℃时有向α-草酸亚铁转变的趋势,80 ℃时β相开始向α相转变,但动力学上因扩散缓慢,样品仍以β-草酸亚铁为主,90 ℃陈化时可得到纯相α-草酸亚铁。Andre[11]认为在90 ℃陈化条件下才能完全完成β-草酸亚铁到α-草酸亚铁的相转变。因此,在后续实验中,除特殊提到外,样品均为90 ℃陈化合成的α-草酸亚铁。
由Fe-H2O系电位-pH图可知:在理想情况下,pH≤6且电势较低的区域为Fe2+的稳定区[12]。本文研究了pH对材料结构的影响。
图2所示为不同pH条件下合成草酸亚铁的XRD图谱。从图2可见:样品均为单相的α-草酸亚铁,但是随着pH的增大,衍射峰的峰强逐渐下降,峰形变宽,这种现象在(002),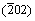 ,(202),(402)和(204)等晶面的衍射峰均有体现。以(202)为例,当pH=1时,衍射峰的半高宽WFWHM=0.192;当pH=6时,WFWHM=0.239。说明当pH比较低时,产物的结晶性比较好,结晶完善。当pH值增大时,由于部分Fe2+会被氧化为Fe3+,导致结晶性变差。实验过程的实际溶液体系与理想的平衡溶液体系有一定偏离,故当pH≤6时,仍有部分Fe2+会被氧化为Fe3+,随着pH增加,Fe2+越来越容易被氧化,Fe3+浓度增加,导致草酸亚铁的结晶度降低。
,(202),(402)和(204)等晶面的衍射峰均有体现。以(202)为例,当pH=1时,衍射峰的半高宽WFWHM=0.192;当pH=6时,WFWHM=0.239。说明当pH比较低时,产物的结晶性比较好,结晶完善。当pH值增大时,由于部分Fe2+会被氧化为Fe3+,导致结晶性变差。实验过程的实际溶液体系与理想的平衡溶液体系有一定偏离,故当pH≤6时,仍有部分Fe2+会被氧化为Fe3+,随着pH增加,Fe2+越来越容易被氧化,Fe3+浓度增加,导致草酸亚铁的结晶度降低。
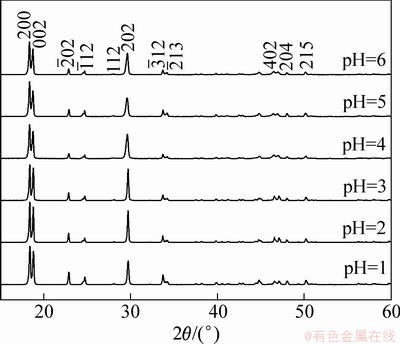
图2 不同pH值条件下合成草酸亚铁的XRD图谱
Fig. 2 XRD patterns of FeC2O4・2H2O prepared at different pH values
图3所示为不同温度下陈化7 h合成草酸亚铁的红外吸收光谱。从图3可见:水合草酸盐类的2个明显的特征吸收峰,即位于3 350 cm-1波段处结晶水的吸收峰和位于1 630 cm-1波段处C=O的反对称拉伸振动吸收峰。位于1 320 cm-1波段处的谱峰为O―C―O的拉伸振动吸收峰,820 cm-1波段处的谱峰为O―C―O的弯曲振动吸收峰,因此,采用该方法合成草酸亚铁的特征峰与水合草酸盐类的特征吸收峰一致。不同相结构的红外吸收光谱主要差别在于1 000~400 cm-1波段处的谱峰,可能是Fe―O的振动吸收峰[13-14],由于pH及温度的差别,导致结构及其结晶性有差异,Fe―O的振动吸收峰也会发生变化,这与XRD分析结果相一致。
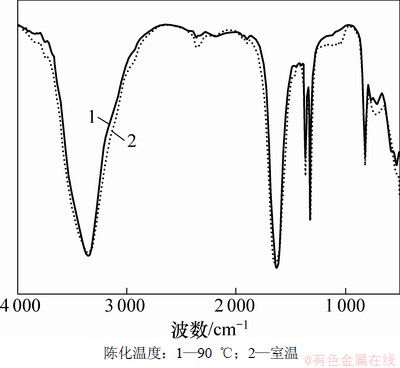
图3 α-草酸亚铁和β-草酸亚铁的红外吸收光谱图
Fig. 3 Infrared spectra of α- and β-ferrous oxalate dehydrates
2.2 草酸亚铁的激光粒度分析
化学合成产物粒度大小及其分布取决于结晶过程中形核率和晶体长大速率的控制。形核率是指单位时间、单位体积内所形成的晶体数目,可表示为
I∝nvexp(-Q/(kT)) (1)
其中:n为单位体积内的原子数;v为原子振动频率,Q为原子扩散激活能;k为波尔兹曼常数;T为热力学温度[15]。
溶液中晶体生长的主要过程是:结晶物质从溶液本体内向晶体表面扩散,结晶物质在表面上吸附,最简单的粒子沿表面移动,最后以某种方式嵌入晶格,形成粗大的结晶体[16]。晶体长大速率可用其晶面的平均线生长速率表示,可用公式表达为
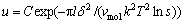 (2)
(2)
其中:C为常数,l为单层厚度,vmol为溶解物质的分子体积,k为波尔兹曼常数,T为绝对温度,δ为溶液过饱和度,s为溶液过饱和系数[15]。
在硫酸亚铁浓度为2 mol/L,分散剂与水的体积比为2:1,pH=3,搅拌速度为1 500 r/min,陈化温度为90 ℃的条件下分别以2种加料方式合成草酸亚铁粉末,其平均粒径如表1所示。
表1 不同进料方式合成草酸亚铁粉末的平均粒径
Table 1 Particle size of FeC2O4・2H2O prepared in different ways of material feeding

从表1可知:硫酸亚铁往草酸溶液中滴加得到的草酸亚铁粉末粒径更小。这是因为:按照这种进料方式,母液中C2O42-浓度远大于Fe2+加入量浓度,不断加入的Fe2+会与C2O42-生成Fe(C2O4)x-2(x-1)(x=1~3),直到达到饱和后,才会逐渐电离生成草酸亚铁,开始成核和晶粒长大的过程,晶粒生长速度较慢,故晶粒尺寸较小;而草酸溶液往硫酸亚铁溶液中进料时,母液中的Fe2+浓度远大于草酸加入量浓度,故不经过生成Fe(C2O4)x-2(x-1)的过程,立即反应生成草酸亚铁,成核和晶粒长大的速率都很快,而且在这种生成环境下易造成粒子团聚,从而导致粉末的平均粒径偏大,并且导致先后形成的颗粒粒度很不均匀[17]。
在硫酸亚铁浓度为2 mol/L,分散剂与水的体积比为2:1,pH=3,陈化温度为90 ℃的条件下,以硫酸亚铁往草酸溶液中进料的方式以不同的搅拌速度合成草酸亚铁粉末,平均粒径见表2。
从表2可知:随搅拌速度的增加,粉末的平均粒径减小。溶液的搅拌可以加速离子的扩散,起到降低扩散激活能Q的作用;另一方面,由于机械振动的作用,溶液中会出现浓度的波动,因而可产生高过饱和区,也可加速扩散。因此,随着搅拌强度的增加,晶体的形核速率显著加快。搅拌强度对于晶体生长速率也有一定影响,搅拌强度大时可减小晶粒表面附近液体层厚度,从而可在一定程度上加快晶体生长速度,但是,搅拌强度对晶体生长速率的影响过程很复杂,当搅拌强度达到一极限值后,晶体生长速率不再发生变化[15]。故搅拌强度对晶体形核速率的影响较大,因此,晶体的平均粒径随搅拌速度的加快而减小。此外,搅拌可以阻止聚集体的生成,保证晶体生长条件一致,有利于生成大小均匀的粒子。并且搅拌的过程中还会伴随着相应的物理磨碎过程,随着搅拌强度的提高,团聚的二次大颗粒被磨碎的也多,这个过程也会导致粉末粒度下降。
在硫酸亚铁浓度为2 mol/L,分散剂与水的体积比为2:1,搅拌速度为1 500 r/min,陈化温度为90 ℃的条件下,以硫酸亚铁往草酸溶液中进料的方式控制不同的pH值合成草酸亚铁粉末,其平均粒径结果如表3所示。
表2 不同搅拌速度下合成草酸亚铁粉末的平均粒径
Table 2 Particle size of FeC2O4・2H2O prepared using different stirring speeds
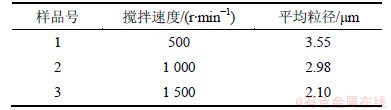
表3 不同pH下合成草酸亚铁粉末的平均粒径
Table 3 Particle size of FeC2O4・2H2O prepared using different pH values
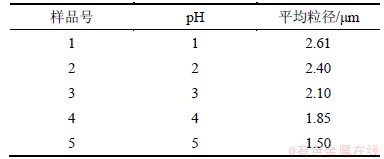
由表3可知:随着pH的增大,样品平均粒径逐渐下降。这是因为:随着pH值的增加,母液中电离出的游离C2O42-越多,进入的Fe2+会先与C2O42-形成配合物,游离C2O42-越多,配合物达到饱和而电离出草酸亚铁所需的时间越长,故草酸亚铁晶粒的生长过程就越缓慢,晶粒就越小;并且随着pH的增加,母液体系的黏度增大,减小粒子间相互碰撞聚集的几率,从而降低产物平均粒度。
在硫酸亚铁浓度为2 mol/L,搅拌速度为1 500 r/min,pH=3,陈化温度为90 ℃的条件下,以硫酸亚铁往草酸溶液中进料的方式加入不同量的分散剂合成草酸亚铁粉末,其平均粒径结果如表4所示。分散剂可以降低样品颗粒的表面张力,防止颗粒的沉降和颗粒间的聚集,抑制晶粒间的结合,使晶粒生长缓慢,从而可有效降低粉末粒径。
表4 不同分散剂与水的体积比下合成草酸亚铁粉末的平均粒径
Table 4 Particle size of FeC2O4・2H2O prepared using different volume ratios of dispersant and water
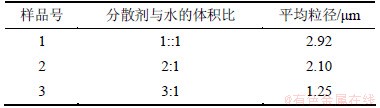
表5所示为采用不同浓度的硫酸亚铁溶液合成草酸亚铁粉末的平均粒径。从表5可见:随着硫酸亚铁溶液的浓度增大,粉末的平均粒径呈现逐渐下降的趋势;溶液浓度大时,反应体系的过饱和度就越大,晶体生长速率降低。此外,溶液浓度大,反应体系的黏度相应增大,溶液体系中粒子运动受到阻碍,粒子间相互碰撞聚集的几率减小,因此,晶体的平均粒径也会减小[16]。
2.3 草酸亚铁形貌分析
图4(a)所示为采用草酸溶液往硫酸亚铁溶液中滴加的进料方式合成草酸亚铁的SEM图,从图4(a)可见:晶粒呈粗大的圆柱状,晶粒表面不光滑,附着一些形貌不规则的纳米颗粒,粗颗粒柱体长度约为10 μm,直径为3~5 μm,该结果与激光粒度测试结果一致,说明按照此方法合成的草酸亚铁晶粒粗大,颗粒大小不均匀,并且结晶性不好。图4(b)所示为pH为5、无分散剂、室温陈化合成草酸亚铁的SEM图。图4(b)中草酸亚铁为0维超细晶粒的疏松团聚态,这些0维纳米颗粒粒度在100~300 nm之间。图4(c)所示为在pH为3、加有分散剂、60 ℃陈化的条件下合成草酸亚铁的SEM图。从图4(c)可见:草酸亚铁晶粒呈一维杆状,晶粒间无明显团聚,颗粒表面光滑并且粒度均匀,这些一维杆的长度在1.5~3 μm之间,直径为200~400 nm。图4(d)所示为pH为3、无分散剂、90 ℃陈化的条件下合成草酸亚铁的SEM图。由图4(d)可见:晶粒呈现二维片状,晶粒表面光滑,二维片的长为1~2 μm,宽为500~800 nm,片的厚度为50~100 nm。因此,相比较于pH=5,pH=3时合成草酸亚铁的结晶性更好,这与XRD分析结果一致。从图4可见:分散剂的添加可以有效减少晶粒间的团聚。
表5 不同硫酸亚铁浓度下合成草酸亚铁粉末的平均粒径
Table 5 Particle size of FeC2O4・2H2O prepared using different FeSO4 contents
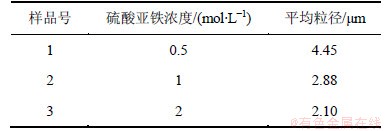

图4 不同条件下合成草酸亚铁的SEM图
Fig. 4 SEM images of FeC2O4・2H2O prepared at different conditions
α-草酸亚铁属于单斜晶系,其Fe2+和草酸根基团形成一种无限链状结构,如图5所示。草酸根基团在这里起到四齿配位基的作用,Fe2+另外与2个H2O相连,形成了1个变形的FeO6八面体环境,这些链状结构平行于b轴,并形成规则排列的层状结构,层状结构垂直于c轴。故α-草酸亚铁沿着b轴有着明显的各向异性结构,草酸亚铁链状结构的1维特性对草酸亚铁某些晶面的各向异性生长有很大的影响。β-草酸亚铁有类似的链状结构,只是这些链状结构在形成层状时发生了混排,使得层与层之间的氢键重新排列[18-19]。
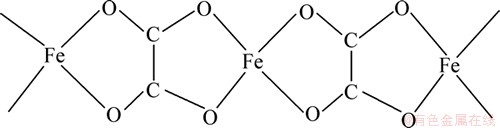
图5 FeC2O4・2H2O中Fe2+和草酸根基团无限链状结构的示意图
Fig. 5 Schematic drawing of infinite chain arrangement present in Fe2+ oxalates of FeC2O4・2H2O
室温陈化时,因温度比较低,晶体生长比较缓慢,所以,得到颗粒细小的0维超细颗粒,晶体的各向异性不明显;随着温度的升高,晶粒某些晶面的生长速率明显增加,至60 ℃陈化时,晶体呈现明显的1维杆状结构,这些1维杆状晶粒的生成,可能和草酸亚铁1维链状结构有着密切的联系;至90度陈化时,因温度升高,一些小晶粒容易被较大晶粒吸附,而且晶粒生长速率也增加,故晶粒逐渐变得粗短,形成粗大的片状,而且片状结构可能和草酸亚铁链状结构形成的层状结构相关。
3 结论
(1) 不同陈化温度合成草酸亚铁的结构不同,室温陈化时得到纯相β-草酸亚铁,随着陈化温度的升高,逐渐向α-草酸亚铁转变,至90 ℃陈化时,得到纯相α-草酸亚铁。
(2) 采用硫酸亚铁往草酸溶液里面滴加的进料方式,合成草酸亚铁的平均粒度更小更均匀;硫酸亚铁浓度的升高、搅拌速度的增大和分散剂比例的增大,均有利于减小草酸亚铁粉末平均粒径。
(3) 随着pH升高,草酸亚铁的平均粒径逐渐减小,但其结晶性随pH的升高逐渐变差。
(4) 采用常规原料,通过控制合成条件,可以合成0维超细颗粒状、1维杆状和2维片状的草酸亚铁。
参考文献:
[1] Doherty C M, Caruso R A, Smarsly B M, et al. Colloidal crystal templating to produce hierarchically porous LiFePO4 electrode materials for high power lithium ion batteries[J]. Chemistry of Materials, 2009, 21(3): 2895-2903.
[2] 张宝, 李新海, 朱炳权, 等. 低温合成LiFePO4/C正极材料及其电化学性能[J]. 中南大学学报: 自然科学版, 2006, 37(3): 505-508.
ZHANG Bao, LI Xinhai, ZHU Bingquan, et al. Low temperature synthesis and electrochemical properties of LiFePO4/C cathode[J]. Journal of Central South University: Science and Technology, 2006, 37(3): 505-508.
[3] CHEN Weimin, QIE Long, YUAN Lixia, et al. Insight into the improvement of rate capability and cyclability in LiFePO4/polyaniline composite cathode[J]. Electrochimica Acta, 2011, 56(6): 2689-2695.
[4] Konarova M, Taniguchi I. Synthesis of carbon-coated LiFePO4 nanoparticles with high rate performance in lithium secondary batteries[J]. Journal of Power Sources, 2010, 195(11): 3661-3667.
[5] 彭春丽, 沈超, 张宝, 等. 溶液浓度对前躯体FePO4・xH2O及LiFePO4性能的影响[J]. 中南大学学报: 自然科学版, 2010, 41(5): 1668-1673.
PENG Chunli, SHEN Chao, ZHANG Bao, et al. Effect of solution concentration on FePO4・2H2O precursor and performance of LiFePO4[J]. Journal of Central South University: Science and Technology, 2010, 41(5): 1668-1673.
[6] NI Jiangfeng, Morishita M, Kawabe Y, et al. Hydrothermal preparation of LiFePO4 nanocrystals mediated by organic acid[J]. Journal of Power Sources, 2010, 195(9): 2877-2882.
[7] CHENG Fuquan, WAN Wang, TAN Zhuo, et al. High power performance of nano-LiFePO4/C cathode material synthesized via lauric acid-assisted solid-state reaction[J]. Electrochimica Acta, 2011, 56(8): 2999-3005.
[8] GAO Fei, TANG Zhiyuan, XUE Jianjun. Effects of different iron sources on the performance of LiFePO4/C composite cathode materials[J]. Journal of University of Science and Technology Beijing, 2008, 15(6): 802-807.
[9] 卢阳, 童庆松, 翁秀燕, 等. LiFePO4/C的制备电化学性能研究[J]. 福建师范大学学报: 自然科学版, 2009, 25(4): 68-71.
LU Yang, TONG Qingsong, WENG Xiuyan, et al. Synthesis and electrochemical performance of LiFePO4/C samples[J]. Journal of Fujian Normal University: Natural Science Edition, 2009, 25(4): 68-71.
[10] 刘旭恒, 赵中伟. 碳源和铁源对LiFePO4/C材料的制备及性能的影响[J]. 中国有色金属学报, 2008, 18(3): 541-545.
LIU Xuheng, ZHAO Zhongwei. Effects of carbon source and iron source on preparation and performance of LiFePO4/C[J]. The Chinese Journal of Nonferrous Metals, 2008, 18(3): 541-545.
[11] Andre A. Synthesis of magnetite nanoparticles by thermal decomposition of ferrous oxalate dehydrate[J]. J Mater Sci, 2008, 43(15): 5123-5130.
[12] 李荻. 电化学原理[M]. 北京: 北京航空航天大学出版社, 1999: 88-98.
LI Di. Electrochemistry principles[M]. Beijing: Beijing University of Aeronautics and Astronautics Press, 1999: 88-98.
[13] ZHOU Weiwei, TANG Kaibin, ZENG Suyuan, et al. Room temperature synthesis of rod-like FeC2O4・2H2O and its transition to maghemite, magnetite and hematite nanorods through controlled thermal decomposition[J]. Nanotechnology, 2008, 19(6): 1-9.
[14] Gabal M A. Non-isothermal decomposition of NiC2O4-FeC2O4 mixture aiming at the production of NiFe2O4[J]. Journal of Physics and Chemistry of Solids, 2003, 64(8): 1357-1385.
[15] 哈姆斯基 E B. 化学工业中的结晶[M]. 古涛, 叶铁林, 译. 北京: 化学工业出版社, 1984: 37-44, 66-69.
Xam X B. Crystallization in chemistry industry[M].GU Tao, YE Tie-lin. Beijing: Chemistry Industry Press, 1984: 37-44, 66-69.
[16] 彭爱国, 贺周初, 余长艳, 等. 高纯超细草酸亚铁的制备研究[J]. 精细化工中间体, 2008, 38(4): 56-58.
PENG Aiguo, HE Zhouchu, YU Changyan, et al. Study on the preparation of ultrafine high-purity ferrous oxalate[J]. Fine Chemical Intermediates, 2008, 38(4): 56-58.
[17] 唐红辉. LiFePO4正极材料及其改性研究[D]. 长沙: 中南大学材料科学与工程学院, 2006: 20-21, 39-40.
TANG Hong-hui. LiFePO4 cathode material and its modification[D]. Changsha: School of Material Science and Engineering. Central South University, 2006: 20-21, 39-40.
[18] Maria C, D’Antonio, Alejandra W, et al. Spectroscopic investigations of iron(Ⅱ) and Iron(Ⅲ) oxalates[J]. J Braz Chem Soc, 2009, 20(3): 445-450.
[19] WANG Dewei, WANG Qihua, WANG Tingmei. Controlled synthesis of mesoporous hematite nanostructures and their application as electrochemical capacitor electrodes[J]. Nanotechnology, 2011, 22(13): 1-12.
(编辑 邓履翔)
收稿日期:2012-06-29;修回日期:2012-10-16
基金项目:科技部创新基金资助项目(10C26224302621)
通信作者:苏玉长(1964-),男,湖南冷水江人,教授,从事新型锂离子电池材料和新型稀土功能材料的研究;电话:0731-88830785;E-mail:ychsu@csu.edu.cn

