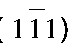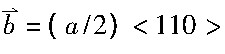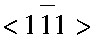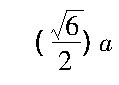网络首发时间: 2016-07-13 09:51
稀有金属 2017,41(03),233-238 DOI:10.13373/j.cnki.cjrm.XY15031906
铝合金中刃型位错与合金元素相互作用的分子动力学模拟研究
刘明辉 肖伟 王建伟 王立根
北京有色金属研究总院有色金属材料制备加工国家重点实验室
摘 要:
采用分子动力学方法和修正的嵌入原子势 (MEAM) , 系统地研究了Mg, Si, Fe 3种元素与Al刃型位错的相互作用。研究结果表明, 在铝合金中, Mg, Si, Fe 3种合金元素都易于偏聚到位错周围, 且Mg和Si与位错的相互作用强于Fe, 展现了更良好的固溶强化效果。另外, 为了阐明时效强化时铝合金中元素聚集对材料力学性能的影响, 进而研究了不同尺寸的Mg团簇与位错的相互作用, 研究发现铝合金中的大尺寸的团簇与位错相互作用更强烈, 与位错相互吸引, 产生明显的钉扎作用, 阻碍位错的运动。本研究从原子尺度分析了铝合金中不同种类元素 (Mg, Si, Fe) 和不同尺寸的元素团簇与位错的相互作用, 阐述了时效过程中合金元素对材料强化的影响, 为今后实验和新型铝合金的开发提供了理论指导。
关键词:
铝合金 ;MEAM ;团簇 ;刃型位错 ;
中图分类号: TG146.21
作者简介: 刘明辉 (1989-) , 男, 河北河间人, 硕士研究生, 研究方向:材料模拟仿真, E-mail:mhliuhust@hotmail.com;; 王立根, 教授, 电话:010-82241124, E-mail:lg_wang1@yahoo.com;
收稿日期: 2015-03-25
基金: 国家自然科学基金项目 (51204022) 资助;
Atomistic Simulation on Interaction of Edge Dislocation with Alloy Elements in Aluminum Alloys
Liu Minghui Xiao Wei Wang Jianwei Wang Ligen
State Key Laboratory for Fabrication and Processing of Nonferrous Metals, General Research Institute for Nonferrous Metals
Abstract:
Molecular dynamics simulations with the modified embedded atom method ( MEAM) were used to study the interaction between Al edge dislocation and Mg, Si, and Fe dopants in the alloy. The calculated results showed that Mg, Si, Fe atoms tended to gather around the dislocation in the Al alloy. Compared with Fe, the interactions of the edge dislocation with Mg and Si were stronger, so Mg and Si had a better solid solution strengthening effect in this alloy. Furthermore, in order to clarify the influence of alloy elements aggregation on the mechanical properties of aluminum alloys during aging process, the interaction of the dislocation with the Mg clusters was studied. It was found that larger clusters in aluminum alloys had a stronger interaction with the dislocations. The larger cluster could obviously draw the dislocations and inhibited their movement, resulting in a better pinning effect. The present work analyzed the interaction of dislocations with Mg, Si, Fe elements and element clusters with various sizes in aluminum alloys at the atomic scale. Based on these results, we further shed light on the strengthening effects of alloying elements in aluminum alloys during the aging process, which would provide a theoretical guidance for future experiments and the development of new aluminum alloys.
Keyword:
aluminum alloys; MEAM; clusters; edge dislocations;
Received: 2015-03-25
随着社会的不断进步, 汽车工业得到了迅猛发展, 汽车在给人们生活带来便利的同时, 也带来了诸如环境污染、能源过度消耗和交通安全隐患等一系列问题。汽车的节能减排是汽车工业继续发展急需解决的问题。汽车轻量化作为节能减排的一个有效手段
[1 ]
, 吸引了大批材料科研人员专注于轻量化结构材料的研究, 而铝合金凭借其可靠的安全性, 易于回收利用等优势得到了更多的关注, 其中铝合金中合金元素对其性能的影响是研究的一个重点方向。
在铝合金中, 合金元素Mg, Si, Fe等以置换固溶体的方式固溶于铝晶体中, 同时铝合金中存在大量位错等缺陷, 在铝合金的塑性变形过程中, 合金元素或形成的析出相与位错相互作用, 阻碍位错的运动, 从而起到强化的作用
[2 ,3 ,4 ,5 ,6 ,7 ,8 ]
。位错的运动与晶体的塑性变形密切相关
[9 ]
, 因此人们对位错行为的研究倾注了很大的力量。为了更直观的展现位错的结构, 研究者建立了许多位错模型进行理论分析, 并取得了很多研究成果:如PeierlsNabarro位错模型, Foreman位错模型等
[10 ,11 ]
。这些模型已经突破了弹性力学研究范围, 初步处理了位错芯的结构, 为今后微观尺度研究位错滑移奠定了基础。
计算模拟技术是材料研究的重要方法, 并且已经运用到合金的位错研究中。Yasi等
[12 ,13 ]
运用第一原理和嵌入原子势 (EAM, embedded atom method) 对镁合金中的位错芯进行了研究, 并将不同方法的模拟计算结果与实验结果进行了对比, 发现合适的EAM势计算的结果与实验结果相符合。为了进一步研究合金中固溶元素对合金塑性变形性能的影响, Xiao等
[14 ]
已经将EAM运用于研究镁合金中Al原子、及Al团簇对塑性变形的影响, 验证了Al在Mg合金中固溶强化作用, 并阐述了团簇尺寸对位错运动的影响。
本文中, 利用基于原子间相互作用的经验势MEAM系统地研究了Mg, Si, Fe 3种元素与Al刃型位错的相互作用, 从原子尺度上阐明了在Al合金时效处理过程中合金元素的固溶强化对材料力学性能的影响。
1 实验
在本文中, 将针对Al合金中合金元素的固溶强化效果进行原子尺度的模拟。模拟过程如下:首先分别研究了单个的Mg, Si, Fe原子与刃型位错的相互作用;其次, 针对Mg团簇, 研究了不同尺寸的颗粒与刃型位错的相互作用。
研究中, 采用Peierls-Nabarro模型建立位错结构;使用含有Al, Mg, Si, Fe的MEAM势
[15 ]
描述原子间的相互作用 (面心立方的铝合金晶体的晶格常数a=0.405 nm) ;模拟软件选用LAMMPS (large-scale atomic molecular massively parallel simulator) 。位错滑移过程中, 常见的基面刃型位错在Al合金塑性变形过程中发挥着重要的作用。如图1所示, 选择的刃型位错基滑移面为
为了描述位错与合金元素的相互作用, 定义结合能Eb 为晶体中位错与合金元素在当前较近距离状态下体系的总能量相对于位错与合金元素相距无限远时体系总能量差。在模拟过程中, 随着合金元素与位错距离的逐渐增加, 结合能收敛且小于0.001 e V, 可认为此时合金元素与位错的距离足够远, 取为参考值 (即结合能Eb =0) , 位错与合金元素其他状态下的结合能由此确定。根据定义, 结合能的正负表示合金元素与位错芯的相互作用趋势, 结合能为正值 (负值) 意味着体系中合金元素与位错相互排斥 (吸引) ;结合能的大小即绝对值决定着两者相互作用的剧烈程度。
2 结果与讨论
2.1 铝位错与Mg, Si, Fe单个原子的相互作用
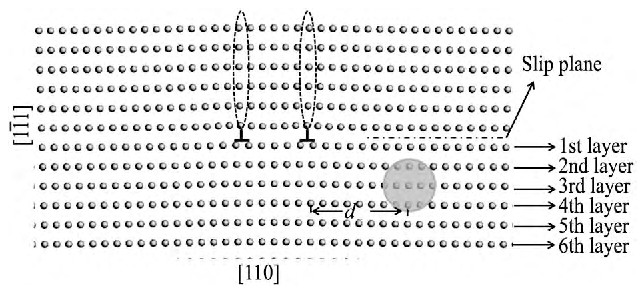
首先, 对Al刃型位错结构进行了弛豫优化, 并得到了它的几何特征。如图1所示, 一个单位位错被分为两个部分位错 (由“┷”表示) , 且两个部分位错 (虚线椭圆部分标出的多余原子面) 之间的宽度为3b (b为刃型位错的柏氏矢量的模) 。虚直线所在面为体系的滑移面, 在此面以上的部分原子排列密集, 为压缩状态, 滑移面以下的部分原子排列相对稀疏, 呈拉伸状态, 此模型与其他研究中
[16 ,17 ]
建立的铝合金位错模型一致。
图1 Al刃型位错模型Fig.1 Model of Al edge dislocation
本文主要针对Mg, Si和Fe 3种不同单原子的固溶强化作用进行原子尺度的模拟。为了准确模拟单个原子与位错的相互作用, 将合金元素作为替代原子被依次放置于优化后原胞的系列位置。考虑到刃型位错芯结构的复杂性以及原胞的对称性, 合金元素位置标记如下:滑移面上的压应力区或滑移面下的拉应力区;距离滑移面的原子层数n, 即Y方向。由于紧邻滑移面两侧的原子介于拉应力区和压应力区之间, 原子受力环境复杂, 因此仅选取了距离滑移面2, 3, 4, 15层的原子进行模拟;相距位错的距离d, 即X方向。从近到远, 分别选取了距离右不全位错芯为0b, 1b, 2b, 4b, 6b, 8b, 10b, 15b, 20b, 25b的位置进行模拟。
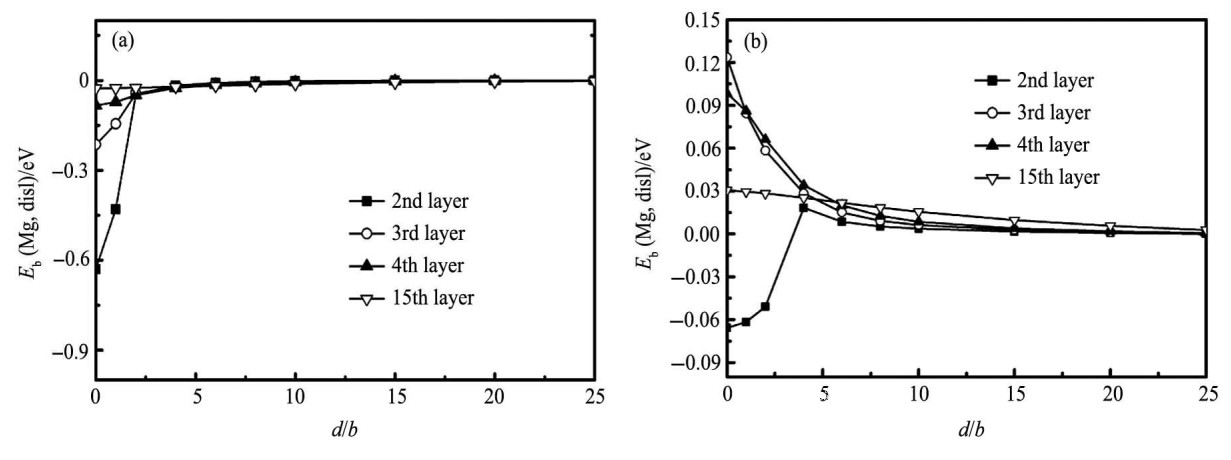
对于单个原子与位错的相互作用, 仅考虑原胞Z方向的长度L=20的情况。在图2中, 分别列出了位于滑移面以下和滑移面以上区域的单个Mg原子与位错结合能与Mg原子位置之间的规律。从图2 (a) 中, 可以发现当Mg原子置换位于滑移面以下的Al原子时, 结合能全部为负值, 并且结合能随两者之间距离d增大而逐渐减小, 根据结合能定义可以知道, Mg原子与位错之间相互吸引, 且易于偏聚到位错周围。对于相同的距离d, Mg原子所处的原子层越靠近滑移面则结合能越大。当Mg原子与位错沿X方向距离d=3b时, 结合能急剧下降, 接近于0, 此时Mg原子与位错芯之间作用几乎消失。当Mg原子位于滑移面以上位置时, 如图2 (b) 所示, 除第二层原子外结合能几乎为正值, 这表明Mg原子与位错相互排斥, 且当Mg原子远离位错时, 结合能降低, 位错与位错芯的相互作用变弱, 最终作用消失。这意味着在压应力区, Mg原子不易偏聚到位错周围。滑移面上的第二层原子, 由于离位错芯区及拉应力区较近, 受力仍比较复杂, Mg原子在这层某些位置上表现出很弱的吸引力, 但并不能改变在压应力区, 溶质Mg原子与位错相互排斥的结论。
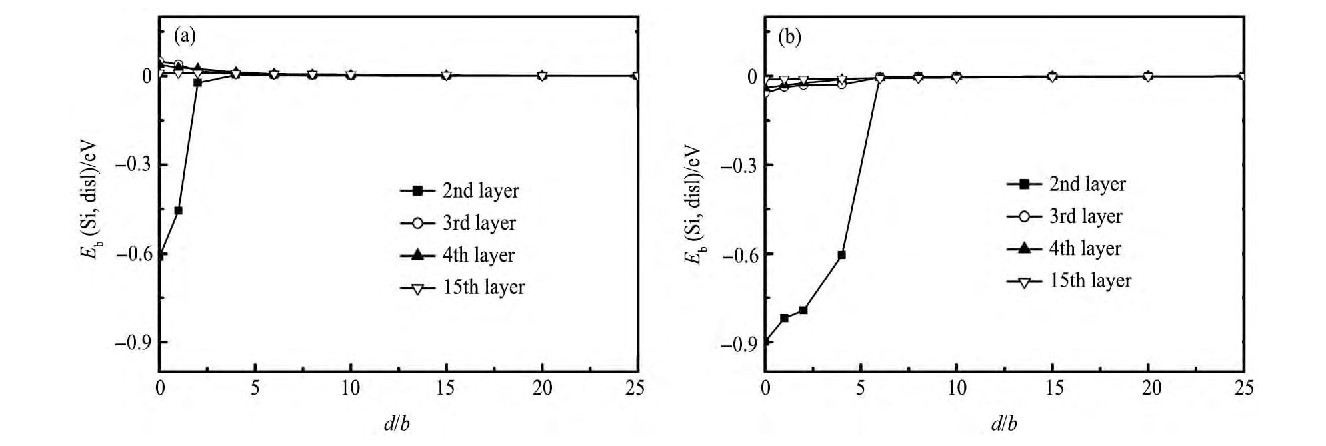
另外, 还研究了铝合金中其他两种添加元素Si和Fe与位错的相互作用情况, 分别由图3和4给出。在图3 (a) 中, 可以发现当Si原子位于滑移面以下的拉应力区第二原子层时, Si原子与位错相互吸引;但从第三层之后, Si原子与位错之间表现为相互排斥。位错芯区及滑移面的存在增加了滑移面下第二层原子与位错相互作用的复杂性。随着距离的增加, Si原子与位错相互作用也逐渐变弱并消失。当Si位于滑移面以上的压应力区原子层时, 见图3 (b) , 位错与合金元素Si原子表现为相互吸引, 且随着两者之间距离减小, 相互作用越来越强烈。这意味着Si在压应力区容易偏聚到位错周围。
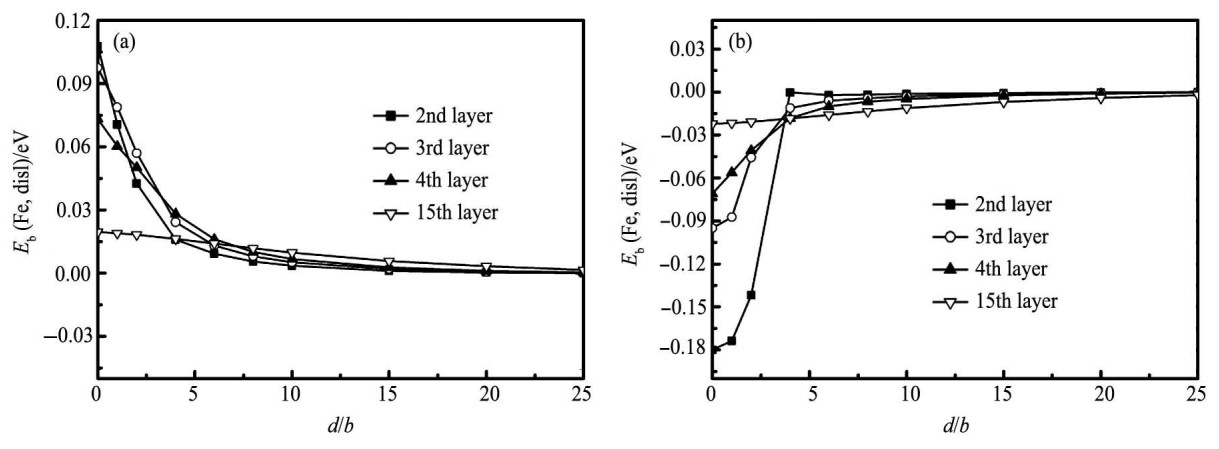
图4中, 我可以发现Fe原子与位错的相互作用如下:当Fe原子置换滑移面以下的Al原子时, 位错与Fe原子之间表现出相互排斥的作用, 它不仅随着原子层远离滑移面而减弱, 而且在同一原子层上随着Fe原子离位错距离d的增加而逐渐消失;当Fe原子位于滑移面以上部分时, 位错与Fe原子之间则相互吸引, 吸引力伴随着Fe原子远离位错而逐渐减弱。研究表面Fe原子易于在压应力区偏聚到位错周围。
图2 位错与Mg之间结合能曲线Fig.2 Binding energy (Eb) between dislocation and dissolved magnesium atom
(a) Magnesium atom being in tensile region below slip plan; (b) Magnesium atom being in compressive region above slip plan
图3 位错与Si之间结合能曲线Fig.3 Binding energy (Eb) between dislocation and dissolved silicon atom
(a) Silicon atom being in tensile region below slip plan; (b) Silicon atom being in compressive region above slip plan
图4 位错与Fe之间结合能曲线Fig.4 Binding energy (Eb) between dislocation and dissolved iron atom
(a) Iron atom being in tensile region below slip plan; (b) Iron atom being in compressive region above slip plan
通过以上研究, 可以得到如下结论:Mg原子易于偏聚到拉应力区域, 而Si和Fe原子易于偏聚到压应力区域。这可能主要由合金元素的尺寸效应决定。分别考察了Al, Mg, Si, Fe 4种原子的半径, 这里的原子半径指的是单质晶体中最近两原子间距的一半。经考察可知, Mg原子半径为0.160nm, 大于Al原子半径0.143 nm, 因此Mg位于拉应力区域更舒适, 引起的晶格畸变小;Si原子半径为0.134 nm, Fe原子半径0.127 nm, 两者半径均小于Al原子半径, 则位于压应力区域更舒适。此外, 还观察到, 合金元素Mg, Si与位错的相互作用强于Fe与位错的相互作用, 这表明Mg, Si更容易偏聚到位错周围, 产生较好的固溶强化效果。当材料发生塑性变形引起位错发生滑移时, Mg, Si的阻碍作用更强。因此在铝合金中添加Mg和Si元素相比于添加Fe元素更能提高铝合金的强度。
2.2 铝位错与Mg团簇的相互作用
以往的研究表明, 铝合金中添加的合金元素在时效初期会发生聚集而形成团簇
[18 ,19 ]
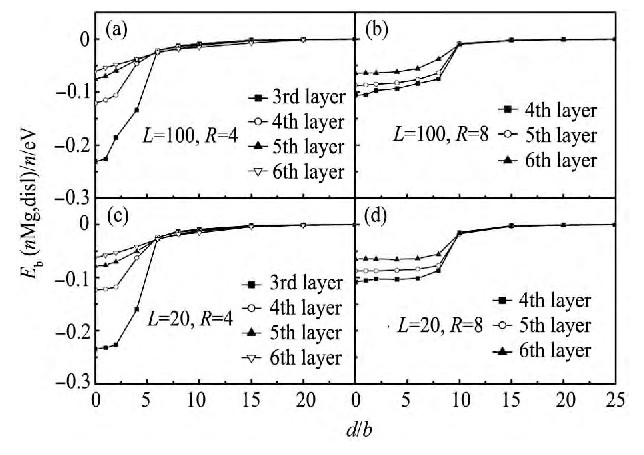
, 合金元素形成团簇后与位错的相互作用可能会有所改变。前文中, 通过对不同合金元素与位错的相互作用的研究, 证实了位于拉应力区域的Mg原子更易偏聚到位错周围。因而, 接下来, 特别研究了位于拉应力区域的Mg团簇与位错的相互作用。为了模拟不同尺寸的Mg团簇与位错的相互作用, 选取了两种尺寸的Mg团簇, 分别为含有19个Mg原子半径为0.4 nm的球形团簇, 以及含有141个Mg原子半径为0.8 nm的球形团簇。团簇离位错的距离由中心原子距离位错的距离d标识。针对每种尺寸的团簇, 又分别考虑了它们与不同长度位错的相互作用, 即L=20和L=100, 不同的L代表体系中两个团簇的距离或不同浓度的团簇。图5中 (a~d) 分别列出了4种不同条件下, 团簇中平均每个Mg原子与位错结合能的变化。
图5 (a) 和 (c) 分别表示出了尺寸较小的Mg团簇在不同浓度时平均每个Mg原子与位错的结合能变化趋势, 图5 (b) 和 (d) 分别表示了尺寸较大的Mg团簇在不同浓度时平均每个Mg原子与位错的结合能变化趋势。从图5可以看出, 团簇中平均每个Mg原子对位错的结合能与单个Mg原子对位错的结合能变化趋势相同, 但是团簇尺寸的变化会影响团簇与位错相互作用的范围以及平均每个原子与位错结合能大小。尤其是作用范围, 通过比较可以发现较大尺寸的团簇 (R=0.8 nm) 与位错距离约为d=10b才会衰减到0, 在距离小于10b时, 团簇与位错之间一直存在较强的相互作用;相较而言, 当团簇尺寸较小时 (R=0.4 nm) , 团簇与位错相互作用范围约为d=6b, 作用距离明显减小。而对于平均每个Mg原子与位错结合能与团簇尺寸大小关系不是很明显, 但由于较大尺寸的团簇含有较多的Mg原子, 则就整个团簇而言, 它与位错相互作用更为强烈。
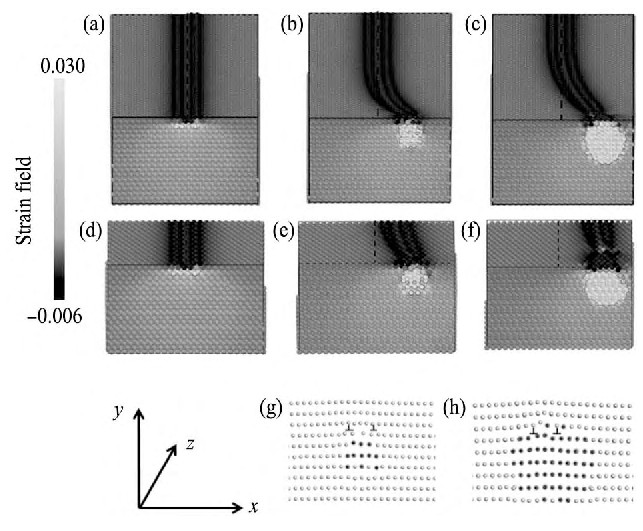
为了更直观地描述位错与团簇的相互作用, 在图6中绘制出了体系中平均局域应变场, 场函数定义为εi = (1/12) Σj (rij -r0 ij ) /r0 ij , 用于表示原子周围的应变状态, 其中原子j为原子i的所有最近邻原子, rij 为当前体系中原子i和原子j之间的距离, r0 ij 为完整的铝晶体中两个最近邻Al原子之间的距离。在图6中, 由于对称性只显示了Z方向的一半长度;同时, 上平面选取到位错芯区。从而位错线和团簇平均局域应变场的分布情况在图6中可由不同的颜色得以分辨。黑色带状区域为两条不全位错所在位置, 而亮灰色区域为Mg团簇所在位置。图6 (a~c) 绘制的是当L=100即合金中Mg团簇浓度较低时, 不同尺寸的Mg团簇与刃型位错的相互作用情况, 由于位错长度较长, 此时位错线没有发生整体的偏移, 但是靠近团簇的位错部分发生了弯曲, 当团簇尺寸较大时, 见图6 (c) , 弯曲程度更大, 这意味着位错与团簇之间的弹性相互作用更强, 即塑性变形阻力更大。通过对比图6 (d~f) 可以看出, 当L=20即合金中Mg团簇浓度较高时, 合金中小尺寸团簇的存在会使位错整体发生偏移, 见图6 (e) , 但偏移幅度较小;当合金中存在尺寸较大的Mg团簇时, 见图6 (f) , 位错会发生大幅度的整体偏移。图6 (g) 和 (h) 分别表示了图6 (e) , (f) 两图状态下位错芯部的结构, 灰色和黑色圆点分别代表铝原子和镁原子。通过对比可以看出, 大尺寸团簇与位错芯相交的区域, 两个不全位错芯有明显相互靠近的趋势, 见图6 (h) , 这意味着团簇的钉扎会引起位错的芯区结构发生变化。
图5 位错与团簇之间平均每个镁原子的结合能Fig.5 Binding energy (Eb) for per magnesium atom between dislocation and magnesium clusters
(a, c) Smaller (R=0.4 nm) ; (b, d) Larger (R=0.8 nm) magnesium clusters at (a, b) lower (L=100) ; (c, d) Higher concentrations (L=20)
图6 位错和团簇周围的局域应变场示意图Fig.6Local strain field around dislocation core and cluster with lower (a~c) and higher (d~f) concentrations
(a, d) No magnesium cluster; (b, e) Smaller magnesium cluster; (c, f) Larger magnesium cluster; (g) and (h) being atomic structure around dislocation core as in cases of (e) and (f) , respectively
3 结论
主要研铝合金塑性变形过程中, 溶质原子以及团簇与位错的相互作用。首先, 通过研究Mg, Si和Fe元素在铝合金晶体中与位错的相互作用, 了解到由于尺寸效应, Mg易于偏聚到位错拉应力区;Fe, Si易于偏聚到位错压应力区。通过结合能曲线可以发现Mg, Si与位错相互作用强于Fe, 具有更好固溶强化效果。其次, 研究了不同尺寸的Mg团簇与位错的相互作用。研究发现大尺寸的团簇更容易吸引位错, 并产生明显的钉扎作用。本研究从原子尺度形象地描绘了铝合金中合金元素的时效强化过程, 为今后选取合适的固溶强化元素提供了理论参考。
参考文献
[1] Ma M T, Ma L X.Application and technology prospect of aluminum alloy in automotive lightweight[J].New Material Industry, 2008, (9) :4. (马鸣图, 马露霞.铝合金在汽车轻量化中的应用及其前瞻技术[J].新材料产业, 2008, (9) :43.)
[2] Totten G E, Mackenzie D S.Handbook of Aluminum:Vol.1:Physical Metallurgy and Processes[M].Boca Raton:CRC Press, 2003.58.
[3] Vissers R, Van Huis M A, Jansen J, Zandbergen H W, Marioara C D, Andersen S J.The crystal structure of theβ'phase in Al-Mg-Si alloys[J].Acta Materialia, 2007, 55 (11) :3815.
[4] Lodgaard L, Ryum N.Precipitation of dispersoids containing Mn and/or Cr in Al-Mg-Si alloys[J].Materials Science and Engineering:A, 2000, 283 (1) :144.
[5] Gupta A K, Lloyd D J, Court S A.Precipitation hardening in Al-Mg-Si alloys with and without excess Si[J].Materials Science and Engineering:A, 2001, 316 (1) :11.
[6] Zhen L, Kang S B.The effect of pre-aging on microstructure and tensile properties of Al-Mg-Si alloys[J].Scripta Materialia, 1997, 36 (10) :1089.
[7] Jaafar A, Rahmat A, Hussain Z, Zainol I.Effect of Mg, Si and Cu content on the microstructure of dilute6000 series aluminium alloys[J].Journal of Alloys and Compounds, 2011, 509 (35) :8632.
[8] Wang G, Yang H X, Jiao M W, Wei Y S, Li H, Gong Z F.Development and application of aluminum foam material in automobiles[J].Chinese Journal of Rare Metals, 2015, 39 (7) :660. (王刚, 杨红新, 焦孟旺, 魏元生, 李贺, 贡泽飞.泡沫铝在汽车上的开发应用[J].稀有金属, 2015, 39 (7) :660.)
[9] Taylor G I.The mechanism of plastic deformation of crystals.Part I.Theoretical[J].Proceedings of the Royal Society of London.Series A, Containing Papers of a Mathematical and Physical Character, 1934, 145 (145) :362.
[10] Nabarro F R N.Dislocations in a simple cubic lattice[J].Proceedings of the Physical Society, 1947, 59 (2) :256.
[11] Foreman A J, Jaswon M A, Wood J K.Factors controlling dislocation widths[J].Proceedings of the Physical Society.Section A, 1951, 64 (2) :156.
[12] Yasi J A, Hector L G, Trinkle D R.First-principles data for solid-solution strengthening of magnesium:from geometry and chemistry to properties[J].Acta Materialia, 2010, 58 (17) :5704.
[13] Yasi J A, Nogaret T, Trinkle D R, Qi Y, Hector L G, Curtin W A.Basal and prism dislocation cores in magnesium:comparison of first-principles and embedded-atom-potential methods predictions[J].Modelling and Simulation in Materials Science and Engineering, 2009, 17 (5) :055012.
[14] Xiao W, Zhang X, Geng W T, Lu G.Atomistic study of plastic deformation in Mg-Al alloys[J].Materials Science and Engineering:A, 2013, 586:245.
[15] Jelinek B, Groh S, Horstemeyer M F, Houze J, Kim S G, Wagner G J, Moitra A, Baskes M I.Modified embedded atom method potential for Al, Si, Mg, Cu, and Fe alloys[J].Physical Review B, 2012, 85 (24) :245102.
[16] Zhang X, Lu G.Calculation of fast pipe diffusion along a dislocation stacking fault ribbon[J].Physical Review B, 2010, 82 (1) :012101.
[17] Lu G, Tadmor E B, Kaxiras E.From electrons to finite elements:a concurrent multiscale approach for metals[J].Physical Review B, 2006, 73 (2) :024108.
[18] Marioara C D, Andersen S J, Jansen J, Zandbergen H W.Atomic model for GP-zones in a 6082 Al-Mg-Si system[J].Acta Materialia, 2001, 49 (2) :321.
[19] Huang C G, Yi J, Gao Y J.Microstructure evolution of nano-cluster in Al-Mg-Si-Zn alloy in early stage of aging[J].Journal of Guangxi University:Nat.Sci.Ed., 2010, 35 (5) :831. (黄创高, 易杰, 高英俊.Al-Mg-Si-Zn合金时效早期纳米团簇的演变[J].广西大学学报:自然科学版, 2010, 35 (5) :831.)