���ù�����λ���Ʊ���ϸLiMn2O4��������
������1,2, ��Ծ��1, ������3
(1. ���ϴ�ѧ ��ĩұ������ص�ʵ����,���� ��ɳ, 410083;
2. �й���ѧԺ �Ϻ�ϵͳ����Ϣ�����о���,�Ϻ�, 200050;
3. ��ɳ��Ԫ�²��Ϲɷ�����˾,���� ��ɳ, 410100)
ժҪ: ���ù�����λ��Ӧ���Ʊ������a�����������LiMn2O4,������λ��ϼ������ͷ�Ӧ�¶ȶ����ղ�������ࡢ�ȱ�����������͵����ܵ�Ӱ������о����о��������:���ù�����λ�����Ʊ���ϸLiMn2O4��ĩ,��λ������ǰ������550��ʱ����12h,��ĩ������ϸС,������0.06~0.50��m�ķ�ĩռ82.10%;��ĩ��ƽ������Ϊ138.12nm,�ȱ����Ϊ10.15m2/g,������ò����,�ڲ��кܶ���;���ϵ��״γ䡢�ŵ�������ֱ�Ϊ126.0mA��h/g��124.2mA��h/g,�״γ�ŵ�Ч��Ϊ98.6%,��15��ѭ����,���ϵ�����������Ϊ 91%��
�ؼ���: ������λ��ѧ;�⾧ʯLiMn2O4;��ϸ��ĩ;�缫����
��ͼ�����:TM911 ���ױ�ʶ��:A ���±��: 1672-7207(2005)03-0390-06
Preparation of ultrafine LiMn2O4 cathode materials
by solid state coordination method
CHEN Li-bao1,2, HE Yue-hui1, TANG Yi-wu3
(1. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China;
2. Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Sciences, Shanghai 200050, China;
3. Changsha Liyuan Novel Materials Limited Company, Changsha 410100, China)
Abstract: The solid state coordination method was used to synthesize ultrafine LiMn2O4 powder. The influences of the volume fraction of citric acid and the calcining temperature on the purity, specific surface area, particle size and electrochemical properties of LiMn2O4 powders were investigated. The ultrafine LiMn2O4 powder with an average particle size of 138.12nm and a specific surface area of 10.15m2/g is obtained when the initial materials is calcined at 550�� for 12h. The powders with the particle size in range of 0.06-0.5��m account for 82.10%, and the powder show regular morphology and porous internal structure. The first charge and discharge specific capacity of the powders is 126.0mA��h/g and 124.2mA��h/g, respectively, the Coulomb efficiency is 98.6%, and the capacity remains 91% after 15 cycles.
Key words: solid state coordination method; spinel LiMn2O4; ultrafine powder; cathode material
Ŀǰ,��������ӵ�������IJ�����Ҫ�в�״LiCoO2,LiNiO2�ͼ⾧ʯLiMn2O4����ȻLiCoO2�������Ͼ����Ʊ����ռ�,�����ܺõ��ŵ�,����,����ϡ�н���,�����ж�,��������ʹ��DZ��;���⾧ʯLiMn2O4������������ṹ�ȶ�,�Ҿ����Ʊ�����,�ɱ���,��,�ɻ��յ��ŵ�,����Ϊ�缫���ϵ��о��ȵ㡣
�Ʊ��⾧ʯ��LiMn2O4�������ϵķ����ܶ�,��Ҫ�и��¹��෨[1]�����ڽ��շ�[2,3]�����սᷨ[4]��Pechini��[5,6]����������[7,8]���ܽ�������[9,10]����Һ��[11]�ȡ��缫���ϵĵ绯ѧ����ǿ�������ڷ�ĩ�����Ⱥ���ò��Ϊ��ߵ缫���ϵij䡢�ŵ����ܺ�ѭ������,B.J.Hwang��[12]������������ĩ�����缫���о����о��������,������������ɢ����,�ȱ������,������ɢ·����,��ɢ���ʽϿ�,��ʹLi+��������������Ч��Ƕ����ѳ�,ʹ����ĩ����Һ�ĽӴ�������,���������Ӵ��ݡ�Ŀǰ,���õ��ȹ��෨�ɹ��ϳ�������LiCoO2�����������[13]�����������������ĩ[14]������ZnO��NiO��ĩ[15]�ȡ�
1 ʵ�鲿��
1.1 ʵ�鷽��
��������������ﮡ������̺���λ��ϼ������ᰴ�������,���մ��в�����ĥ1h,������ɵ���ɫ��״����λ�������м��塣Ȼ��,���м�����70~80��ʱ��ո���4h,�õ���ɫ���ɿ�״�����ǰ���塣��ǰ�����ڿ�����������300~800��ʱ����12h,��¯��ȴ�õ�����LiMn2O4��
1.2 ���ϵı���
������λ������ǰ����ĺ������չ�����Nicolet-740FT-IR�����Dzⶨ;����/���ȷ������Ϻ���ƽ������������WRT-3P��������ƽ�Ͻ���;�����ձ�RIGAKU D/MAX-3A��Cu��K������X����������,�Բ��ϵľ���ṹ�����о�;��������MICRO-PLUS�ͼ����������ȷ����Dzⶨ���ϵ����ȷֲ�; ��������QUANTACHROME��˾������MONOSORB�ͱȱ�������Dzⶨ����ıȱ����;�ñ�������������˾������ԭ�����շֹ��ȼ�,�ⶨ������﮵ĺ���;�����ձ�JEOL��˾������JSM-5600LV��ɨ����������۲��ĩ����ò��
1.3 �����ܲ���
�����Ƶõ�LiMn2O4��ĩ����Ȳ�ں�ճ�����������8��1��1��Ϻ�,��������ˮ�ͷ�ɢ����ͪ,��ͣ�����Ͼ���,Ȼ������ˮ��,���Ƴ�Լ30��m��ĵ缫��Ĥ������Ĥ��120��ʱ��ո���3h,�õ������缫Ƭ���ý����������,Celgard2400Ϊ��Ĥ,PC+DMC(�����Ϊ1��1)+1mol/L LiPF6�Ļ��Һ�����Һ,��������յ�����������װ��ء�
�����ܲ������人�����������˾������LAND CT2001A�Ͳ���ϵͳ�Ͻ���,ʹ�ü�����ɼ����ݡ����Ե����ܶ�Ϊ0.1C,��ѹ��ΧΪ3.00~4.35V��
2 ��������
2.1 ��λ������ǰ������Ʊ�
������Ϊ1��2��3������ﮡ������̺����������������ĥʱ,������ɿ���״�����䳱,��Ϊ�ۺ�ɫ�ſ�;��������ĥʱ,������ſ�����ϡ,���ų�Ũ�ҵĴ�����ζ,����Ϊ�Դ�����ɫ����״�м��塣
�������γ�sp3�ӻ��������(Li+)�����γ�d2sp3�ӻ���������(Mn2+)�������������γ���λ������[16],����ĥ�����з���������λ��ѧ��Ӧ:
C6H8O7��H2O+Mn(Ac)2��4H2O ��
C8H10O9Mn��xH2O + HAc�� + (5-x)H2O; (1)
C6H8O7��H2O+LiNO3 ��C6H7O7Li��
xH2O+HNO3+(1-x)H2O; (2)
C8H10O9Mn��xH2O+LiNO3 ��
C8H9O9MnLi��xH2O+HNO3�� (3)
�����̺�����������ĥ�����з�����λ��ѧ��Ӧ,Mn2+�������ᷢ����Ӧ(1),�γɵ�����λ������C8H10O9Mn��xH2O,����Li+������Ӧ(2),���в���Li+�μӷ�Ӧ(3)���ڸ���ʱ,��λ�������м���ų������ˮ����,���ᷢ���ֽ�,�м���ת��Ϊ������ɵ���λ������ǰ���塣
�Ը�������õ���λ������ǰ������к������չ����,������ͼ1��ʾ����������ĺ������չ������,ǰ������1400~1600cm-1������,���������εIJ��Գ����������մ��ͶԳ����������մ�,���Ƿֱ�λ��1384cm-1��1596cm-1��,��2�����մ��������ε����������״�,˵��Mn2+��Li+�������ᷢ������������,�γ����������Ρ�
2.2 ����/���ط���
ͨ�������̺�������IJ��ȷ�����֪,�����̺���������250����ǰ�����˶����ˮ���Ⱥ��ڻ����ȷ�Ӧ����������280��ʱ��ʼȼ�շ���,��������250�濪ʼ����ȼ�ա�������Ϊ1��2��3������ﮡ������̺�����������ĥ,������õ�����λ������ǰ���������/���ȷ���(TG/DTA)������ͼ2��ʾ��
![]()
1��ǰ����; 2��������
ͼ 1 ��λ������ǰ�����������ĺ������չ���
Fig. 1 Infrared spectra of coordination
compound and citric acid
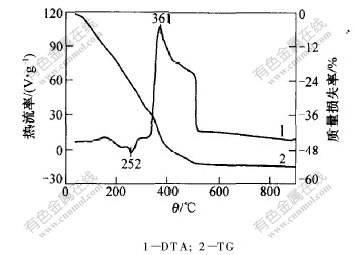
ͼ 2 ��λ������ǰ��������غͲ��ȷ���
Fig. 2 TG/DTA analysis for precursor of
coordination compound
��ͼ2���Կ���,��20~340���ʧ������Ϊ��ǰ�����д��ڽ��ˮ,�ڵ��¸���ʱ,δ��ȫ��ȥ,�ڽϸߵ��¶������ѳ�����DTA������,��252����������Ӧ����ˮ���ȷ塣��Լ340��ʱ,TG���ߵ�ʧ��������,��DTA������Ҳ��ʼ���ַ��ȷ�,˵����340�濪ʼ����������������������ʼ����ȼ��,�ų���������,ͬʱ�⾧ʯ���LiMn2O4��ʼ�γɡ����¶ȴﵽ504���,TG���ߺ�DTA���߶�����ˮƽ������Ʒ����X�������������֪,��Ʒ�Ǵ��ļ⾧ʯ��LiMn2O4��
2.3 ��λ���������Բ����Ӱ��
��������ΪMn2+��Li+����λ��ϼ�,�京���Խ��������Ƿ���ȫ�γ��������������á�����,ǰ�����ڱ��չ�����,����������������ȼ�շų�������,��ʹ�γɼ⾧ʯ��LiMn2O4��ͬʱ,�ų�������CO2��ˮ��,ʹ�����ö�����ɡ����,������������Բ���Ľṹ�����ܶ���Ӱ�졣
����ͬ�����Ƚ�����ﮡ������̡���������,�õ���ǰ������550��ʱ����12h,�õ��IJ���������ͼ3��ʾ����ͼ3���Կ���, ��n(Li+)��n(Mn2+)��n������Ϊ1.0��2.0��1.0ʱ,���ղ����к���Mn2O3�����ࡣ����������������,�������Ӳ�����ȫ�γɹ��������,Li+��Mn2+û����ȫ���;�����ڱ����¶Ƚϵ�,ʱ��϶�,�����ṩ�������ӳ�����ɢ������,���,��ȱ��������Mn2O3�����ࡣ��n(Li+)��n(Mn2+)��n������Ϊ1.0��2.0��(1.5~5.0)ʱ,���ղ��ﶼΪ���⾧ʯ��LiMn2O4�������������ʱ,�ڱ��չ�����ȼ�շų��������ɴٽ��⾧ʯ����γ�,��ѡ��n(Li+)��n(Mn2+)��n������Ϊ1.0��2.0��3.0��Ϊʵ���ԭ����ȡ�
![]()
n(Li+)��n(Mn2+)��n������: a��1.0��2.0��1.0;
b��1.0��2.0��1.5; c��1.0��2.0��2.0; d��1.0��2.0��3.0;
e��1.0��2.0��4.0; f��1.0��2.0��4.5; g��1.0��2.0��5.0
ͼ 3 ��ͬ�������������ò����XRDͼ��
Fig. 3 XRD patterns of lithium manganese
oxide with various amount of citric acid
2.4 �����¶ȶԲ����Ӱ��
ͨ�����ȷ�����֪,��310��ʱ�⾧ʯ��LiMn2O4��ʼ�γɡ���n(Li+)��n(Mn2+)��n������Ϊ1.0��2.0��3.0���õ�ǰ����,�ڲ�ͬ�¶��±���12h,�Բ�����X���߽��о���ṹ���������,�����ͼ4��ʾ���ɼ�,��350���400��ʱ���յIJ����X����ͼ����,���ּ⾧ʯ�͵�LiMn2O4���������,�������������ԵĿ�������,����ǿ��Ҳ�ϵ͡���˵������ľ��β�����,�����̶Ȳ��ߡ�ͬʱ,Ҳ������ƶ﮵�Mn3O4�����ࡣ������Ϊ�����¶ȹ���,����������ɢ�����,��ɢ�����ȡ��������¶ȸ���500��ʱ,�⾧ʯ�͵�LiMn2O4����������ü���,����ǿ��Ҳ�ϸ�,˵���õ��˽ᾧ��ȫ�ļ⾧ʯLiMn2O4����500���550��ʱ���յõ��IJ����Ǵ��ļ⾧ʯ��LiMn2O4,û�г����ø��¹��෨��500��ʱ��Ӧ�õ���LiMnO2,Li2MnO3��Mn2O3�������ࡣ��Щ������500~1000�涼���ȶ�[17]��ͨ��ԭ�����չ��ײⶨ��֪,��550��ʱ����12h�IJ�����﮺���Ϊ3.79%,������﮺���(3.84%)����,��˵���ڵ����±����û����ʧ���ٽ������/���ȷ�����֪,500~550���DZ��յ�����¶ȷ�Χ�����¶ȸ���600��ʱ,�������ֳ�����������Mn3O4��Mn2O3��������Ϊ���ű����¶ȵ�����,һ����⾧ʯLiMn2O4��������������,��һ����,��������������ʹ����Mn��O������,Mn3O4��Mn2O3�Ӽ⾧ʯLiMn2O4�ṹ���������,�γ������ࡣ
![]()
�����¶�/��: a��350; b��400; c��500; d��550;
e��600; f��700; g��800; h��900
ͼ 4 ��ͬ�¶��±���12h���ò����XRDͼ��
Fig. 4 XRD patterns of lithium manganese
oxide calcined at various temperatures
�봫ͳ�ĸ��¹��෴Ӧ�����,���ù�����λ������������˷�Ӧ��ı���ʱ��,�����˱����¶ȡ����¹��෴Ӧ��������800�����ϵ��¶��¼�Ъʽ����48h����[18],���ұ����ϸ���������ٶȺ����·�ʽ����Ϊ���෴Ӧ��һ����ɢ���Ʒ�Ӧ,����Ӧ��֮��Ľ�������,��Ӧ�������,�Ӵ����С,�����������֮�����ɢ���ɳ�,���·�Ӧʱ���ӳ��ͷ�Ӧ�¶����ߡ������ù�����λ��Ӧ��,�������ӻ�Ͼ���,�����˽������ӵij�����ɢ,���,��Ӧʱ���,�����¶ȵ͡�
2.5 ���ϵ���������ò����
���ü������䷨�����550��ʱ����12h����Ʒ�������ֲ�,��ͼ5��ʾ��
![]()
1���������; 2���ۻ��������
ͼ 5 LiMn2O4���ϵ������ֲ�
Fig. 5 Distribution of LiMn2O4 particle size
��ͼ5���Կ���,LiMn2O4���ϵ������ֲ���Χ�Ͽ�,�����ֲ�������,������2��������,��ֵ����Ϊ0.23��m��������Χ��0.06~0.50��m�ķ�ĩռ82.10%,˵���ڴ������ºϳɵ�LiMn2O4����Ϊ��ϸ�ۡ������ֲ������Ⱥʹ��ڴ��������Ҫԭ����������:һ����,������λ�������м�����Ƚϴ�,�����õ���ǰ�����DZȽϼ�Ӳ�ı���,��Ȼ�ڱ��պ�������,�Զ���������,������һ����û�еõ���Ч�ط���,���Դ��������;��һ����,���ڷ�ĩ������С,�ȱ�����ϴ�,�����ܸ�,�����žۡ�������Ӳ�ž۵ķ�ĩ,������ɢ,������0.50~0.80��m�ķ�Χ���������������
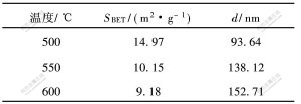
����BET���ⶨ����ıȱ����,����������1��ʾ���ɼ�,��500��ʱ�������ò���ıȱ������14.97m2/g,ͨ����ʽ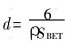 ,��ò��������Ϊ93.64nm�����¶�����550��ʱ,�����ƽ��[CM(22] ������Ϊ138.12nm,�뼤�����������ƽ�������в��������Ϊ��ĩ�������ڲ�������,�����˷�ĩ�ıȱ����,���¼���ó���ƽ��������ֵƫС�����ű����¶ȵ�����,�����������,˵������ͨ�����ںϳ��¶������Ƴ�ϸ�⾧ʯLiMn2O4��ĩ��������
,��ò��������Ϊ93.64nm�����¶�����550��ʱ,�����ƽ��[CM(22] ������Ϊ138.12nm,�뼤�����������ƽ�������в��������Ϊ��ĩ�������ڲ�������,�����˷�ĩ�ıȱ����,���¼���ó���ƽ��������ֵƫС�����ű����¶ȵ�����,�����������,˵������ͨ�����ںϳ��¶������Ƴ�ϸ�⾧ʯLiMn2O4��ĩ��������
�� 1 �ڲ�ͬ�¶�ʱ�������ò���ıȱ����������
Table 1 Specific surface area and particle size of
LiMn2O4 powders calcined at various
temperatures for 12h
��ĩ����ò��ͼ6��ʾ���ɼ�,��ĩ����ϸС,���Ⱦ��ȡ��ɱȱ�������������֪,�����ڲ����ںܶ��϶������������������ڷֽ�����зų�������CO2��H2O,ʹ�����ڲ������ܶ���,�γ���ͨ������,���ַ�ĩ��ò��Բ��ϵĵ绯ѧ�����кܴ�Ӱ�졣

ͼ 6 ��ϸ�⾧ʯLiMn2O4��ĩ��
����ɨ��������Ƭ
Fig. 6 SEM image of ultrafine spinel LiMn2O4
powders calcined at 550�� for 12h
2.6 ���ϵĵ�����
��n(Li+)��n(Mn2+)��n������Ϊ1.0��2.0��3.0�õ�ǰ����,��550��ʱ����12h,�ϳɵļ⾧ʯLiMn2O4��ĩ���ി,����С,ѡ�����ַ�ĩ���״γ䡢�ŵ�ʵ��,���״γ䡢�ŵ�ѭ��������ͼ7��ʾ���ɼ�,�ڳ��ͷŵ������ϸ�����2���Գ��Ժܺõ�ƽ̨�����ƽ̨�����ڵ�ѹΪ4.05V��4.15Vʱ,�ŵ�ƽ̨������4.05V��3.95Vʱ,���ֳ����Ե�LiMn2O4�����䡢�ŵ����ܡ��ŵ��ǰһ��ƽ̨��Ӧ��-MnO2��Li0.5Mn2O4����ƽ��,��һ��ƽ̨��ӦLi0.5Mn2O4��LiMn2O4����ƽ��[19],��ʾ�����õĵ�ѹ�ȶ��ԡ���˵������ӵ�Ƕ����ѳ�����2������,Ƕ�����Ƕ������2���ȶ��ĵ�ѹƽ̨��
���ϵ��״γ�������Ϊ126.0mA��h/g,�״ηŵ������Ϊ124.2mA��h/g,��ŵ�Ч��Ϊ98.6%�����ϵij�ŵ�������ϸ�,������Ϊ��ĩ������С,�ȱ������,һ����ʹLi+����������������Ч��Ƕ����ѳ�;��һ����,ʹ���������ʳ�ֽӴ�,������Li+Ǩ�ơ�
��550��ʱ�ϳɲ��ϵ�ѭ��������ͼ8��ʾ���ɼ�,��15��ѭ����,���ϵķŵ������������113.1 mA��h/g,Ϊ�״ηŵ��������91%,˵�����ϵ�ѭ�����ܽϺá�
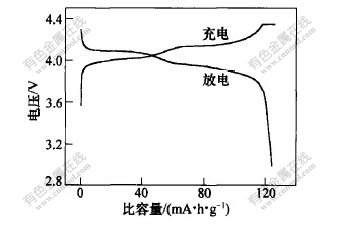
ͼ 7 ��ϸ�⾧ʯLiMn2O4��ĩ
�״γ䡢�ŵ�����
Fig. 7 First charge-discharge curves of
ultrafine spinel LiMn2O4 powder cell
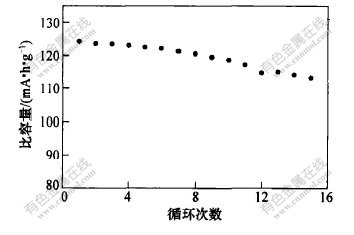
ͼ 8 550��ʱ�ϳɲ��ϵ�ѭ���䡢�ŵ�����ͼ
Fig. 8 Charge-discharge curves of
LiMn2O4 calcined at 550��
3 �� ��
a.���ù�����λ��ѧ��Ӧ�����Ʊ���ϸLiMn2O4��������,�봫ͳ�ĸ��¹��෨���,���б���ʱ���,��Ӧ�¶ȵ�,���ռ��ŵ㡣
b.���ù�����λ��ѧ��Ӧ���ϳ�LiMn2O4,��340��ʱ��λ������ǰ���忪ʼ����ȼ��,�ų�����,�⾧ʯ�͵�LiMn2O4��ʼ�γ�,��504�淴Ӧ������
c.����λ������ǰ����ı����¶ȵ���500��,�����600��ʱ������������,500~550��Ϊ�⾧ʯ��LiMn2O4����Ѻϳ��¶ȡ�
d.��550��ʱ����12h�ϳɵ�LiMn2O4��ĩ,��ò��������,�����ʶ�ṹ,�����ֲ���ΧΪ0.08~0.50��m,�ȱ����Ϊ10.15 m2/g,Ϊ��ϸ��ĩ;��﮺���Ϊ3.79%,������﮺���(3.84%)�����״ηŵ�����Ϊ124.2mA��h/g,ѭ�������ȶ���
�����:
[1] Tarascon J M, Mckinnon W R, Coowar F, et al. Synthesis condition and oxygen stoichiometry effects on Li insertion into spinel LiMn2O4[J]. J Electrochem Soc, 1994, 141(6): 1421-1427.
[2]Aoshima T, Okahara K, Kiyohara C, et al. Mechanism of manganese spinels dissolution and capacity fade at high temperature[J]. J Power Sources, 2001, (97-98): 377-380.
[3]Xia Y, Noguchi H, Yoshio M. A new three-volt spinel Li1+xMn1.5Ni0.5O4 for secondary lithium batteries[J]. J Solid State Chem, 1995, 119(1): 216-218.
[4]�»�, ������, ŷ������. ���ϳ�����ӵ���������ϵĵ�����Ӱ������[J]. ���ܲ���, 2001, 32(4): 385-387.
HAO Hua, LIU Han-xing, OUYANG Shi-xi. Electrochemical influence factors of microwave synthesis cathode material for lithium ion battery[J]. Functional Materials, 2001, 32(4): 385-387.
[5]Liu W, Kowal K, Farington G C. Electrochemical characteristics of spinel phase LiMn2O4 based cathode materials prepared by the Pechini process[J]. J Electrochem Soc, 1996, 143(11): 3590-3596.
[6]Wang G X, Bradhurst M H, Liu H K, et al. Improvement of electrochmical properties of the spinel LiMn2O4 using a Cr doping effect[J]. Solid State Ionics, 1999, 120(4): 95-101.
[7]Barboux P, Tarascon J M, Shokoohi F K. The use of acetates as precursors for the low-temperature synthesis of LiMn2O4 and LiCoO2 intercalation compounds[J]. J Solid State Chem, 1991, 94(1): 185-196.
[8]Haitao H, Peter G B. A 4 V lithium manganese oxide cathode for rocking-chair lithium-ion cells[J]. J Electrochem Soc, 1994, 141(9): L106-L107.
[9]Amine K, Tukamoto H, Yasuda H, et al. Differences in electrochemical behavior of LiMn2O4 and Li1+x��Mn2O4 as 4V Li-cell cathodes[J]. J Electrochem Soc, 1996, 143(5): 1607-1613.
[10]Choy J H, Kim D H, Kwon C W, et al. Physical and electrochemical characterization of nanocrystalline LiMn2O4 prepared by a modified citrate route[J]. J Power Sources, 1999, 77(1): 1-11.
[11]Hwang K T, Um W S, Lee H S, et al. Powder synthesis and electrochemical properties of LiMn2O4 prepared by a emulsion-dying method[J]. J Power Sources, 1998, 74(2): 169-174.
[12]Hwang B J, Santhanam R, LIU D G. Characterization of nanoparticles of LiMn2O4 synthesized by citric acid sol-gel method[J]. J Power Sources, 2001, 97-98: 443-446.
[13]����, Ŭ������, ����Ƽ. ���ȹ��෴Ӧ���Ʊ�����LiCoO2���о�(I)[J]. �ߵ�ѧУ��ѧѧ��, 1999, 20(12): 1847-1849.
XIA Xi, NULI Yan-na, GUO Zai-ping. Studies of nanophase LiCoO2 synthesized by solid state reaction with low heating temperature(I)[J]. Chemical Journal of Chinese Universities, 1999, 20(12): 1847-1849.
[14]ׯ��, ���, ʯ����, ��. ���ȹ��෴Ӧ�Ʊ����������������ĩ[J]. ���ܲ���, 2002, 33(3): 253-255.
ZHUANG Jia, CHI Yan-hua, SHI Jun-ning, et al. A Ce0.25NiFeO3 powder prepared by solid state reaction with low temperature[J]. Functional Material, 2002, 33(3): 253-255.
[15]�ὨȺ, �ֵ���, ֣ﳷ�, ��. ����������������п�ĺϳ��·���[J]. ����ѧѧ��, 1999, 15(1): 95-98.
YU Jian-qun, JIA Dian-zeng, ZHENG Yu-feng, et al. A novel preparation route to nanocrystalline nickel oxide and zinc oxide[J]. Journal of Inorganic Chemistry, 1999, 15(1): 95-98.
[16]������, ����Ǣ, ������. ��λ��ѧ�����̳�[M]. ���: ����ѧ����������, 1990.
ZHU Sheng-yu, ZHOU Yong-qia, SHEN Pan-wen. Coordination chemistry[M]. Tianjin: Tianjin Science and Technology Press, 1990.
[17]�����j, ���, ������, ��. ����ӵ��[M]. ��ɳ: ���ϴ�ѧ������, 2002.
GUO Bin-kun, XU Hui, WANG Xian-you, et al. Lithium ion battery[M]. Changsha: Central South University Press, 2002.
[18]����ʤ, �����. �⾧ʯ��LiMn2O4�缫���ϵ��Ʊ��������о�[J]. ����������ѧѧ��, 2000, 27(1): 65-68.
YANG Wen-sheng, LIU Qing-guo. Synthesis and electrochemical studies of spinel phase LiMn2O4[J]. Journal of Beijing University of Chemical Technology, 2000, 27(1): 65-68.
[19]Chung-Hsin Lu, LIN Shang-wei. Influence of the particle size on the electro-chemical properties of lithium manganese oxide[J]. J Power Sources, 2001(97-98): 458-460.
�ո�����:2004 -09 -12
�����:������(1979-),��,������ɽ��,��ʿ�о���,��������ӵ����ز����о�
������ϵ��: ������,��,��ʿ�о���;�绰:021-62511070-8907; E-mail: libao_chen@163.com

