
李 钊1, 2, 3,姚赢赢1, 2, 3,李博飞4,王利诚1, 2, 3,徐 昊1, 2, 3,种丽娜1, 2, 3,邹建新1, 2, 3
(1. 上海交通大学 材料科学与工程学院,上海200240;
2. 上海交通大学 轻合金精密成型国家工程研究中心,上海 200240;
3. 上海交通大学 氢科学中心,上海 200240;
4. 上海工程技术大学 材料工程学院,上海 201620)
可充镁电池因其高体积比容量、高安全性及原料镁储量丰富等优势,有望成为“双碳”目标下规模化储能技术的优选电化学器件。然而目前可充镁电池还未能实现商业化,这与其存在的一些关键科学问题尚未明晰以及技术瓶颈还未被突破等因素有关。因此,本文从可充镁电池的安全性和能量密度出发,在梳理了可充镁电池发展历史的基础上,总结了可充镁电池器件中电解液、正极和负极材料的研究进展。文中主要介绍了格氏基、磺酸基和硼基电解液对改善镁可逆沉积/溶解和提高电压窗口的重要作用,并对Chevrel相Mo6S8、硫化物和氧化物作为正极材料的储镁机制进行了详细分析。然后对高电压的插层正极材料(尖晶石、层状和聚阴离子化合物)、高容量的转化正极材料(硫、氧和有机分子)和高功率的活性炭正极进行了着重介绍。此外,从电解液/电极界面反应机制着手,对镁、镁合金、铋和锡等金属以及石墨等负极材料进行了梳理分析。最后,本文从材料设计、器件匹配和应用场景角度,对可充镁电池未来商业化的挑战进行了总结和展望。
文章编号:1004-0609(2021)-11-3192-25 中图分类号:O646 文献标志码:A
引文格式:李 钊, 姚赢赢, 李博飞, 等. 可充镁电池:发展、机遇与挑战[J]. 中国有色金属学报, 2021, 31(11): 3192-3216. DOI: 10.11817/j.ysxb.1004.0609.2021-42490
LI Zhao, YAO Yin-yin, LI Bo-fei, et al. Rechargeable magnesium batteries: Development, opportunities and challenges[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(11): 3192-3216. DOI: 10.11817/j.ysxb.1004.0609.2021-42490
自人类文明诞生以来,人类生产力的发展均离不开木材、煤炭和石油等能源物质的使用。这些物质往往直接燃烧产生热能,并进一步转化为机械能和电能,最后将电能通过电缆输运到用电企业和居民家中。但令人忧虑的是,这些天然能源物质资源毕竟有限,不可持续使用且会带来一系列环境问题。发展绿色、低碳、可持续使用的清洁能源将是人类未来发展的唯一且有效的途径。在全球环境治理和应对气候变化的大背景下,我国也提出了“碳达峰”和“碳中和”的“双碳”目标[1-2]。地球上最丰富的清洁能源是太阳能,其衍生产物还有风能和水能等。现有的技术可以将这些清洁能源转化为电能[3],例如,硅基太阳能电池等就可以有效地将太阳能转化为电能,但还需将电能存储起来,以应对夜晚及无光照条件时居民就无法使用电能的问题;一种极为有效的方法便是将电能以电化学反应的方式转化为化学能存储到可充放的电池中[3-4]。
目前,已经大规模应用的可充放电池有铅酸和锂离子电池等[5-6]。其中,锂离子电池具有高能量密度,已经广泛用于便携式电子设备和电动汽车上,但是锂离子电池材料中使用的锂、钴和镍等资源毕竟有限[7-8];同时,铅酸电池存在着低能量密度和环境不友好等问题。因此,对于清洁能源的大规模化存储方面,还需开发新型的可充放电池。
可充放的离子电池的本质是,以离子作为电荷载体在正负极之间的穿梭和固定来完成能量的储存和释放[9]。从热力学层面来看,这种放电和充电反应容易发生;同时在动力学层面,快速的离子反应还能够实现电池快速充放电。除了锂离子(Li+)外,很多研究表明一价的钠离子(Na+)[10-11],二价的镁离子(Mg2+)、钙离子(Ca2+)和锌离子(Zn2+)[12],以及三价的铝离子(Al3+)等均可作为可充放的离子电池,这些离子电池的工作原理在大多数方面都与锂离子电池相似[13]。其中,由于地壳中镁的储量比锂高3个数量级,这极大程度上降低了镁电池原材料的相对成本,并缓解了锂供应不足所带来的诸多问题[14]。
镁一次电池在1943年便已开始商业化,这种水激活的氯化银/镁(AgCl/Mg)电池,以Ag/AgCl为正极和Mg为负极,在水加入后发生放电,电压可达1.6 V (vs Mg),可用于军事和航空航天备用电源[15]。相比而言,可充镁二次电池至今仍处于不断探索中。20世纪70年代开始,陶氏公司的GREGORY等[16]便开展可充镁电池电解液和正极材料的研究工作,但直到AURBACH等[17]在2000年报道的Chevrel相Mo6S8作为可充镁电池正极材料匹配镁金属负极,在有机卤铝酸镁/四氢呋喃电解液中,可以实现>2000次循环,才真正地让可充镁电池进入到人们的视野中。
截至目前,可充镁电池尚未实现商业化,还停留在实验室研究阶段。这与镁电池中存在的一些关键科学问题和技术瓶颈尚未解决有关。因此,本文试图从可充镁电池的能量密度和安全性出发,对可充镁电池电解液、正极材料和负极材料的发展和现状进行系统梳理和总结。同时,着重介绍了电解液/金属镁负极界面反应和正极材料的储镁机制所面临的困难与挑战;最后,强调了材料设计和器件匹配策略,对可充镁电池实现实用化作出总结和展望。
1 可充镁电池的能量密度和安全性
可充镁电池作为能源存储器件,需满足高能量密度和安全使用这两个关键要素。电池的能量密度由比容量与电压的乘积(Ah·kg-1×V=Wh·kg-1)决定,比容量与正负极材料可容纳离子数有关,电压则与正极/电解液和负极/电解液的界面电势差有关。如图1所示,列举了几种金属负极的理论质量和体积比容量以及工作电压(vs SHE (标准氢电极))。镁的理论质量比容量(2205 mA·h/g)低于锂(3860 mA·h/g)[18],但高于钠(1166 mA·h/g)[19]。Mg2+与阴离子O2-或S2-结合时,会转移两个电荷,且Mg2+的半径(0.86 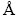 )接近于Li+的半径(0.9 );因此,镁具有更高的理论体积比容量(3833 mA·h/cm3),接近于锂(2046 mA·h/cm3)的两倍,接近于石墨负极的5倍(800 mA·h/cm3)。镁的氧化还原电压(Mg2+/Mg,-2.36 V vs SHE)虽低于锂(Li+/Li,-3.05 V vs SHE),但明显高于铝和锌的氧化还原电压(Al3+/Al,-1.66 V vs SHE;Zn2+/Zn,-0.76 V vs SHE)[20]。
)接近于Li+的半径(0.9 );因此,镁具有更高的理论体积比容量(3833 mA·h/cm3),接近于锂(2046 mA·h/cm3)的两倍,接近于石墨负极的5倍(800 mA·h/cm3)。镁的氧化还原电压(Mg2+/Mg,-2.36 V vs SHE)虽低于锂(Li+/Li,-3.05 V vs SHE),但明显高于铝和锌的氧化还原电压(Al3+/Al,-1.66 V vs SHE;Zn2+/Zn,-0.76 V vs SHE)[20]。
电池充放电循环过程中,锂离子在负极侧沉积时,非常容易产生树枝状或苔藓状的枝晶刺穿隔膜,进而导致电池内短路,发生起火等危险。1989年加拿大Moli公司开发的以锂负极和二硫化钼正极组成的可充锂电池,因锂离子在金属锂负极处无规则的枝晶生长而造成电池隔膜被刺穿,致使电池内部正负极接触而发生电池内短路,造成了多起电池的爆炸事件[21]。20世纪90年代,吉野彰等使用石墨负极取代了锂负极,才实现了电池中使用锂离子的商业化器件[22]。现有研究表明,镁离子在金属镁表面沉积并不会产生枝晶,而是以高度紧密堆积的趋势进行,这也是镁电池使用高比容量的金属镁直接作为负极实现电池安全的最大驱动力之一[23]。使用金属镁作为负极,必须要寻找到合适的电解液来实现镁离子在金属镁表面的可逆溶解和沉积。
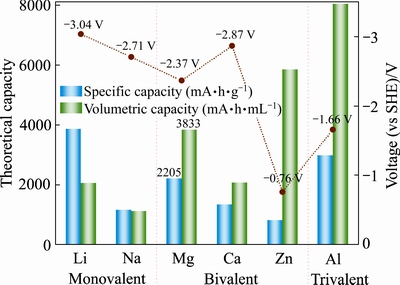
图1 几种金属负极的理论比容量和工作电压
Fig. 1 Theoretical capacities and voltages for various metal anodes
2 可充镁电池电解液
可充镁电池的电解液由溶剂和电解质组成,溶剂需要具有稳定的电压窗口,以保证电解质在金属镁负极表面进行可逆地镁沉积和溶解。在溶剂的选择上,由于镁会与水反应,也会和非水溶剂的酯、砜、酰胺、腈等发生还原反应而生成氧化镁(MgO)等组成的钝化膜,阻碍镁离子的扩散[24]。因此,可充镁电池电解液往往使用醚类溶剂。在电解质的筛选上,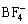 、
、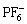 、
、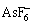 和
和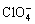 也会在金属镁负极上分解并形成稳定的MgO、Mg(OH)2和MgF2等组成的钝化膜,不仅阻碍镁离子的扩散,还造成镁离子无法可逆地溶解和沉积。因此,诸如格氏基、磺酸基和硼基化合物等电解液由于其相对金属镁的稳定性成为很有前途的选择[25]。
也会在金属镁负极上分解并形成稳定的MgO、Mg(OH)2和MgF2等组成的钝化膜,不仅阻碍镁离子的扩散,还造成镁离子无法可逆地溶解和沉积。因此,诸如格氏基、磺酸基和硼基化合物等电解液由于其相对金属镁的稳定性成为很有前途的选择[25]。
2.1 格氏基电解液
格氏试剂由法国化学家格林尼亚(Grignard)在1900年将有机卤素化合物(卤代烷、活泼卤代芳烃)与金属镁在无水乙醚中反应得到。格氏试剂是含卤化镁的有机金属化合物,分子式为R-Mg-X (R是有机基团,X是卤素),是一种强路易斯碱,具有强烈的亲核性质,可以和某些活泼氢化合物(水、醇、酸等)、CO2、羰基化合物、金属或非金属卤代物等发生反应,称为格林尼亚反应,可用以制备烃类、醇、酮、酸等。格林尼亚也因此发现于1912年获得诺贝尔化学奖[26]。1935年至1942年期间,EVANS等[27-28]研究了几种格氏试剂在乙醚溶剂中的分解电压,乙基溴化镁(EtMgBr)和苯基溴化镁(PhMgBr)的分解电压分别为1.2 V和2.2 V vs Mg。1953年,法国化学家诺尔芒以四氢呋喃(THF)作为溶剂得到了格氏试剂。1973年至1990年期间,三次全球石油危机促使以西方国家为首的全球各国家开始研究电化学储能电池用于替代燃油动力。这期间,陶氏化学公司的GREGORY等[16]提出了以THF为溶剂的乙基氯化镁(EtMgCl)电解液,Cl-相比于Br-具有更强的吸电子特性,易使镁离子以EtMg+的形式在金属镁表面发生镁沉积。同时,他们发现在EtMgCl(1~2 mol/L)中加入少量的强路易斯酸AlCl3(0.1~0.2 mol/L),可以进一步提高镁的沉积性能。后续研究表明,亲电的AlCl3在电解液中容易以EtAlCl4-形式存在,而镁离子则以更有利于沉积反应的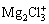 或MgCl+形式存在[29-30]。此外,
或MgCl+形式存在[29-30]。此外,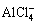 也参与到了镁的溶解反应中,其加入促进了镁可逆地沉积/溶解反应[16]。
也参与到了镁的溶解反应中,其加入促进了镁可逆地沉积/溶解反应[16]。
2000年,AURBACH等[17]使用1 mol/L的正丁基镁(MgBu2)和2 mol/L的乙基氯化铝(AlCl2Et)反应得到丁基-乙基氯酸铝镁(Mg(AlCl2BuEt)2)。如图2(a)所示的循环伏安曲线,Mg(AlCl2BuEt)2的约2.2 V(vs Mg)的电化学窗口明显高于正丁基氯化镁(BuMgCl)和丁苯基硼酸镁Mg(BPh2Bu2)2两种电解液,说明吸电子Cl-的加入有助于扩展有机铝镁酸盐电解液的电化学窗口。同时,Mg(AlCl2BuEt)2具有10-3 S/cm的电导率和100%的镁可逆沉积和溶解的库伦效率。AURBACH等[31-32]后续研究表明,相对较弱的Al—C键在电化学反应中易通过b-H消除反应而断裂。他们在Mg(AlCl2BuEt)2中添加LiCl或四丁基氯化铵对电解液的镁离子电导率具有显著提升作用[32]。紧接着,他们使用不含b-H的芳香烃基团取代烷烃基团,如路易斯酸苯基氯化镁(PhMgCl)和路易斯碱AlCl3以2:1的反应比例生成亲核性的全苯基配合物(APC)电解质[30](见式(1))。
2PhMgCl + AlCl3→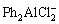 + (1)
+ (1)
如图2(b)所示,相比于Mg(AlCl2BuEt)2 (BEC),APC可达3.2 V(vs Mg)的高电压窗口和2×10-3~ 5×10-3 S/cm电导率,同时具有更窄的过电位和更高的镁可逆沉积/溶解比容量[33]。然后,AURBACH等[34]将LiCl加入APC中,提高了电解液的电导率并再次降低了镁沉积/溶解的过电位(见图2(c));LiCl的加入还增强了电解液在高电压下的稳定性(见图2(c)插图)。
目前来看,APC是比较成熟的格氏基镁电解液。然而,虽然其电压高达3.2 V,但是APC在大于2.2 V时,对铝和不锈钢等非贵金属均具有腐蚀性,这是一个限制APC实用化非常严峻的问题。MULDOON等[35-36]研究表明,腐蚀的发生与二聚体阳离子[Mg2Cl3×(THF)6]+有关。NELSON等[37]使用三苯酚铝(Al(OPh)3)代替AlCl3有效降低了氯化物浓度,缓解了高电压下电解液对不锈钢的腐蚀,但这也降低了溶液中MgCl+或Mg2Cl3+的浓度而不利于镁的沉积。
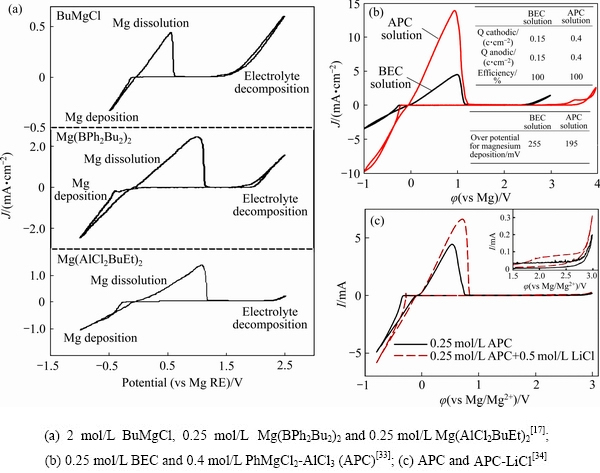
图2 不同电解液中可逆沉积/溶解镁的循环伏安曲线的对比(工作电极,Pt;参比和对电极,Mg)
Fig. 2 Comparison of cyclic voltammograms of different electrolyte solutions in which magnesium being deposited/ dissolved reversibly (Working electrode, Pt; Reference and counter electrodes, Mg)
2.2 含氯的二聚体镁电解液
受格氏试剂中加入AlCl3的启发,DOE等[38]直接使用MgCl2替代格氏试剂和AlCl3直接反应生成了氯化铝镁配合物(MACC),其中MgCl2可以作为路易斯碱与强路易斯酸AlCl3反应,在四氢呋喃(THF)中以2:1的比例形成不含有机金属化合物的MACC,MgCl+和基团偶联的二聚阳离子与阴离子在THF中稳定存在(见式(2))。
2MgCl2+AlCl3→(Mg2Cl3)+×(AlCl4)- (2)
在此基础上,LIU等[39]拓展MACC体系,使用MgCl2–AlPh3和MgCl2–AlEtCl2得到了含有二聚阳离子和相应的阴离子(AlEtCl3)-和(AlClPh3)-。SEE等[40]通过拉曼(Raman)和核磁共振(NMR)光谱以及对分布函数(PDF)分析,探索了MACC中活性与非活性配合物的关系,并证明了Al和Mg具有共依赖性,当[Mg2Cl3·(THF)6]+存在时,游离的Cl-在镁表面富集会有利于镁的沉积。
KIM等[41]将非亲核的六甲基二硅氨基氯化镁(HMDS×MgCl)与AlCl3以3:1比例混合在THF中,其电化学活性中心为[Mg2Cl3×(THF)6]+阳离子和[HMDSn×AlCl4-n]-阴离子,具有3.2 V (vs Mg)的电压稳定性。这种非亲核的镁电解质可以与亲电的硫正极兼容。SEE等[42]将Mg(HMDS)2作为添加剂加入MACC中,改善了镁沉积和溶解[43]。
然而,无论是格氏基电解液APC,还是MACC和HMDS等,都会在MgCl2加入THF中后生成溶剂化的镁二聚体阳离子[Mg2Cl3×(THF)6]+,对铝和不锈钢具有腐蚀作用[36],严重限制了其规模化使用。同时此类电解液的电压稳定性不超过3.2 V vs Mg,因此,对于开发非腐蚀和高电压的电解液的需求十分迫切。
2.3 非腐蚀性的无氯电解液
2.3.1 磺酸基电解液
LOSSIUS等[44]在1995年便发现三氟磺酸镁Mg(CF3SO3)2 (简写为Mg(TFSA)2)可进行镁的可逆沉积和溶解。2014年,HA等[45]报道了双(三氟甲烷磺酰亚胺)镁(Mg[N-(SO2CF3)2]2,简写为Mg(TFSI)2)可以在乙二醇二甲醚(DME)/二乙二醇二甲醚的混合溶剂中可逆沉积镁。0.5 mol/L的Mg(TFSI)2具有5.2×10-3 S/cm的电导率和4 V(vs Mg)高电压稳定性。然而,Mg(TFSI)2的可逆镁沉积/溶解的库伦效率仅为70%[46]。RAJPUT等[47]通过密度泛函理论(DFT)和分子动力学(MD)计算表明,电解液中Mg(TFSI)2容易以MgTFSI+离子对的形式存在,尽管C-S键在TFSI-是稳定的,但在MgTFSI+离子对上是不稳定的。
AURBACH等[48]将MgCl2加入Mg(TFSI)2/DME电解液中,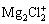 和
和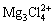 等二聚体镁盐的存在抑制了MgTFSI+离子对的生成,可以有效地将镁沉积过电位降低到200 mV,库伦效率高达99%。MgCl2和AlCl3同时加入,会更进一步提升Mg(TFSI)2的镁的可逆电化学性能[49]。但是溶液中存在的含氯的二聚体镁盐还是具有腐蚀性的。因此,诸如Mg(BH4)2作为电解质添加剂和离子液体作为溶剂添加剂加入Mg(TFSI)2电解液中,来抑制MgTFSI+离子对的生成而来改善镁的可逆沉积[50-52]。
等二聚体镁盐的存在抑制了MgTFSI+离子对的生成,可以有效地将镁沉积过电位降低到200 mV,库伦效率高达99%。MgCl2和AlCl3同时加入,会更进一步提升Mg(TFSI)2的镁的可逆电化学性能[49]。但是溶液中存在的含氯的二聚体镁盐还是具有腐蚀性的。因此,诸如Mg(BH4)2作为电解质添加剂和离子液体作为溶剂添加剂加入Mg(TFSI)2电解液中,来抑制MgTFSI+离子对的生成而来改善镁的可逆沉积[50-52]。
此外,通过添加二甲胺(DMA)等助溶剂,其仲胺官能团提高了THF溶剂对Mg2+的亲和力,促进了Mg(TFSI)2在醚类溶剂中的溶解[53]。紧接着,王春生课题组使用多齿(六齿、三齿和二齿)的甲氧乙基胺螯合剂([-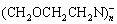 ])来改进DME溶剂[54]。Mg2+与多齿螯合剂的高亲和力使得所组成的溶剂化鞘层比Mg2+(DME)3溶剂化物更稳定,这种较不致密的溶剂化鞘层加快了Mg2+在电解液和电极界面之间的电荷转移,降低了Mg(TFSI)2的过电位,并抑制了电解液与正负极之间的副反应。
])来改进DME溶剂[54]。Mg2+与多齿螯合剂的高亲和力使得所组成的溶剂化鞘层比Mg2+(DME)3溶剂化物更稳定,这种较不致密的溶剂化鞘层加快了Mg2+在电解液和电极界面之间的电荷转移,降低了Mg(TFSI)2的过电位,并抑制了电解液与正负极之间的副反应。
2.3.2 硼基电解液
早在1957年,CONNOR等[55]使用MgBr2和LiBH4制备得到了硼氢化镁(Mg(BH4)2),可以在乙醚中进行电化学镁沉积。1971年,BRENNER[56]实现了Mg在十硼烷镁(MgB10H12×2Et2O)中的电化学沉积。1990年,GREGORY等[16]通过二丁基镁与路易斯酸三丁基硼反应合成了一种电解质(Mg[B(C4H9)4]2),该电解质具有较好的阴离子稳定性,但库仑效率较低。
2012年,MOHTADI等[57]使用Mg(BH4)2溶于DME溶剂中用于可充镁电池,Mg(BH4)2电解质由于具有较高的还原稳定性,可以实现可逆的Mg沉积/溶解;不过电压稳定性仅保持在1.7 V(vs Mg)。接着他们使用高稳定性的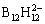 代替
代替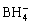 ,然而的弱配位特性导致其不易溶于低极性的醚类溶剂[58]。然后,他们又将MgCl2引入,将电解液的电压提高至3.2 V[58]。最后,他们进一步使用中性的
,然而的弱配位特性导致其不易溶于低极性的醚类溶剂[58]。然后,他们又将MgCl2引入,将电解液的电压提高至3.2 V[58]。最后,他们进一步使用中性的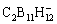 ,合成了碳硼烷团簇Mg(CB11H12)2[59],实现了镁的可逆溶解和沉积,电压窗口高达3.2 V。由于CB11H12–在Mg负极中更为稳定,Mg(CB11H12)2相较于Mg(TFSI)2具有更优异的镁沉积/剥离性能[60]。HAHN等[61]将F–引入,得到Mg(CB11H11F)2,将电压稳定性提升至4.6 Vvs Mg。尽管碳硼烷团簇Mg(CB11H12)2在四乙二醇二甲醚中具有高度可逆的Mg沉积和溶解特性,但是其较高的黏度导致了电池性能的降低。因此,姚彦课题组[62]通过短链的乙二醇二甲醚和长链的二乙二醇二甲醚溶剂混合,制备了具有高效的反应动力学的Mg(CB11H12)2电解液。
,合成了碳硼烷团簇Mg(CB11H12)2[59],实现了镁的可逆溶解和沉积,电压窗口高达3.2 V。由于CB11H12–在Mg负极中更为稳定,Mg(CB11H12)2相较于Mg(TFSI)2具有更优异的镁沉积/剥离性能[60]。HAHN等[61]将F–引入,得到Mg(CB11H11F)2,将电压稳定性提升至4.6 Vvs Mg。尽管碳硼烷团簇Mg(CB11H12)2在四乙二醇二甲醚中具有高度可逆的Mg沉积和溶解特性,但是其较高的黏度导致了电池性能的降低。因此,姚彦课题组[62]通过短链的乙二醇二甲醚和长链的二乙二醇二甲醚溶剂混合,制备了具有高效的反应动力学的Mg(CB11H12)2电解液。
一般来说,较弱的阴离子-阳离子相互作用会有利于阳离子的去溶剂化,进而提高阳离子的电导率[63]。因此,LUO等[64]设计并合成了Mg[B(O2C2(CF3)4)2]2,其中[O2C2(CF3)4]-与B可以进行强配位,因此减弱[B(O2C2(CF3)4)2]-对Mg2+的耦合作用,有利于Mg2+在溶液中的解离。努丽燕娜等[65]设计了一种四(三氟乙醇氧)硼酸镁(Mg[B(Otfe)4]2),弱配位的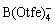 的电解质具有良好的氧化稳定性、高离子电导率和弱腐蚀性。镁可逆沉积/溶解的库伦效率高达99%,过电位低于200 mV,电压窗口超过3 V(vs Mg)。
的电解质具有良好的氧化稳定性、高离子电导率和弱腐蚀性。镁可逆沉积/溶解的库伦效率高达99%,过电位低于200 mV,电压窗口超过3 V(vs Mg)。
尽管Mg(BH4)2直接作为电解液的电压窗口过低,但将其用于镁电解液的添加剂中,有利于促进Mg负极表面和界面的活化。LI等[66]通过电化学和光谱分析表明,Mg(BH4)2添加剂用于烷氧基硼酸镁(Mg[B(hfip)4]2),有助于去除天然氧化层,促进Mg金属镁表面形成固态电解质界面层。WANG等[67]通过对电化学阻抗谱(EIS)和弛缓时间(DRT)分布的分析,确定了Mg、Mg2+与BH4–的相互作用更强,使BH4–在Mg金属表面的电荷转移反应中优先吸附。这同时防止Mg金属和TFSI–之间的不良副反应,从而实现了高度可逆的Mg沉积/溶解。
2.3.3 其他新型电解液
KEYZER等[68]突破了Mg(PF6)2会在金属镁表面生成钝化MgF2,不能用于镁电池电解液的难题,即使用(CH3CN)6配合Mg(PF6)2,在V(THF): V(CH3CN)为1:1的溶剂中可以实现Mg的沉积和溶解,电导率最高可达2.8×10-2 S/cm,具有4 V(vs Mg)的电化学窗口。HERB等[69]将烷氧基铝酸盐作为阴离子,合成了六氟异丙醇铝酸镁Mg[Al(hfip)4]2电解质,也获得了3.5 V高电压稳定性,电导率为6.5×10-3 S/cm。LAU等[70]设计并合成了四氟叔丁基铝酸镁Mg(TPFA)2,其中阴离子TPFA-(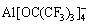 )是高度缺电子的,可以与Mg2+保持弱配位,这提高了其相对金属镁负极的稳定性,是实现3.8 V(vs Mg)高电压窗口的关键。烷氧基硼酸盐阴离子更轻,因此烷氧基硼酸镁(Mg[B(hfip)4]2)也被研究[71]。不过Mg[Al(hfip)4]2和Mg[B(hfip)4]2都是水溶性,在水分存在下会水解,因此在实际应用过程中面临困难。二茂镁(双(η5-环戊二烯基)镁)也被报道[72],但是电压稳定性仅到1.8 V(vs Mg)。
)是高度缺电子的,可以与Mg2+保持弱配位,这提高了其相对金属镁负极的稳定性,是实现3.8 V(vs Mg)高电压窗口的关键。烷氧基硼酸盐阴离子更轻,因此烷氧基硼酸镁(Mg[B(hfip)4]2)也被研究[71]。不过Mg[Al(hfip)4]2和Mg[B(hfip)4]2都是水溶性,在水分存在下会水解,因此在实际应用过程中面临困难。二茂镁(双(η5-环戊二烯基)镁)也被报道[72],但是电压稳定性仅到1.8 V(vs Mg)。
2.4 离子液体
离子液体通常作为溶剂或助溶剂,具有提高电解液稳定性的作用,同时由于其不易燃而提高了电池的安全特性。不过,离子液体的高粘度和溶质阳离子的低迁移数会导致较差的电导率,因而有机醚类溶剂和离子液体的混合被证明可以提高镁离子的电导率[73]。
努丽燕娜等[74]首次报道将离子液体1-丁基-3-甲基咪唑四氟硼酸盐(BMIMBF4)加入到Mg(TFSA)2中。随后,他们又使用BMIMBF4、1-甲基-1-丙基哌啶双(三氟甲磺酰)亚胺盐(PP13TFSI)和Mg(TFSA)2组成混合型离子液体[75],将Mg沉积过电位从400 mV降到200 mV。相比而言,MORITA等[76]将N,N-二乙基-N-甲基-N-(2-甲氧基乙基)氨(三氟甲磺酰)亚胺(DEMETFSI)和双(三氟甲磺酰)亚胺(R-IMTFSI)(R为烷基或烷烃基)加入格氏基电解液中,则优化了Mg的可逆溶解和沉积。
2.5 镁固态电解质
镁液态电解质受限于醚类溶剂的电压窗口窄、含氯电解质具有腐蚀性等问题,跳过镁液态电解质而开发具有更高电压窗口和稳定性的镁固态电解质将是可充镁电池实现实用化的一个不错的选择。事实上,镁固态电解质在1987年便开始被研究,IKEDA等[77]首次报道Mg-Zr-PO4体系镁固态电解质,在400 ℃和800 ℃下电导率分别为2.9×10-5和6.1×10-3 S/cm。2000年,IMANAKA等[78-79]报道了Zr2O(PO4)2,其电导率是Mg-Zr-PO4体系的两倍。不过与锂固态电解质所面临的困境类似,镁固态电解质还需大幅提升室温下Mg2+的电导率。
不同于坚硬的无机固态电解质,固态有机聚合物电解质与颗粒状电极的不规则表面相接触还能保持柔软和一致性界面,可有效地增加Mg2+在固固接触界面处的电导率[80]。由Mg(AlCl2EtBu)2/四聚氰胺/聚偏二氟乙烯(PVDF)组成的凝胶固态电解质[81],室温下的电导率为3.7×10-3 S/cm。Mg(BH4)2-MgCl2在聚四氢呋喃(PTHF)形成的凝胶固态电解质的室温电导率为4.76×10-4 S/cm,在-20 ℃时的电导率为5×10-5 S/cm[82]。
金属有机框架材料由于其开放式骨架特点便于高电荷密度的Mg2+的移动,近些年来被研究用于镁固态电解质。AUBREY等[83]报道以金属有机骨架Mg2(2,5-二氧苯-1,4-二羧酸盐)和Mg2(4,4′-二氧联苯-3,3′-二羧酸盐)为材料,将酚酸镁置换成不饱和金属位,合成了一系列固态镁电解质。在金属有机框架(MOF): Mg(TFSI)2或Mg(OPhCF3)2混合物中,室温下镁电导率最高达到2.5×10-4 S/cm。MINER等[84]开发的Cu基金属多氮唑框架(Cu4(ttpm)2·0.6CuCl2),MOF-MgBr2的离子电导率为1.3×10-4 S/cm。
近些年,Mg(BH4)2作为镁固态电解质的可能性被报道[85-87]。YAN等[86]将中性分子NH3引入Mg(BH4)2中,形成的氢键N—Hd+…-dH—B促进主体晶格间发生了交换,形成Mg2+间隙通道,在80 ℃下镁离子的电导率为3.3×10-4 S/cm。
2.6 小结
在可充镁电池电解液中,Mg2+通常会与溶剂形成缔合更强的溶剂化结构,这对镁离子在电解液和电极/电解液界面传输性能影响很大。已发展成熟的格氏基电解液APC,由于所使用的强路易斯酸AlCl3,能将溶剂化镁变成一价态的MgCl+,使之更易于在电解液中传输和在电极/电解液界面中发生解离,能有效地实现镁沉积。磺酸基电解质Mg(TFSI)2和硼基电解质Mg(CB11H12)2中的弱配位阴离子TFSI-和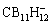 ,通过额外加入Mg(BH4)2等电解质添加剂或多齿配位的胺类螯合剂等助溶剂,来加快镁离子去溶剂化过程,并以缓解界面副反应来促进镁离子的可逆沉积。
,通过额外加入Mg(BH4)2等电解质添加剂或多齿配位的胺类螯合剂等助溶剂,来加快镁离子去溶剂化过程,并以缓解界面副反应来促进镁离子的可逆沉积。
APC、Mg(TFSI)2和Mg(CB11H12)2是镁基电解质中公认的突破性成果,搭配环状的四氢呋喃(THF)或链状的乙二醇二甲醚(DME)等醚类溶剂,在近些年的研究中被广为使用。其中,以APC作为电解质时,多匹配低电压高容量硫族化合物正极材料,Mg(TFSI)2则多使用高电压氧化物正极材料,而Mg(CB11H12)2较适用于有机正极材料。然而,易于获取的APC具有一定腐蚀性,不适用于以不锈钢或铝等作外壳的电池器件,可使用高分子材料如塑料等做电池器件外壳。而Mg(TFSI)2和Mg(CB11H12)2则由于合成步骤较多和价格昂贵,在未来的实用化生产中不得不需要面临降低成本的问题。此外,镁固态电解质将会是突破现有的液态电解液存在电压窗口窄的一个良好的解决方案。研究人员在探索新型镁固态电解质时,不仅仅要考虑室温下镁离子在固态电解质中的电导率提升上,更应把注意力放在固态电解质与正极或负极的固固接触界面上镁离子的迁移率提升。
3 可充镁电池正极材料
商业化的锂离子电池是以锂离子在正极材料中的嵌入/脱出的插层机制实现的能量存储。然而,高电荷密度的二价镁离子与材料主体结构的氧或硫等阴离子会产生较大库伦排斥力,难以进入多数材料的晶格内部。Chevrel相Mo6S8的出现,将人们探索镁插层正极材料的视线再次拉进。
3.1 Chevrel相Mo6S8
层状、尖晶石和橄榄石结构的含锂氧化物都是紧密排列的氧阴离子亚晶格结构,相比而言,Chevrel相是一个独特的“簇”结构。首先通过高温固相反应合成Cu2Mo6S8[88],然后将Cu原子从Cu2Mo6S8中酸浸出[89],便留下带有空腔的Mo6S8,因此Mo6S8在热力学上是亚稳态的。如图3(a)所示,6个Mo在立方体表面形成了八面体,8个S占据角落,形成了空腔[13]。如图3(b)所示,空腔1离Mo原子最远且与Mo6S8共享角落,空腔2和3则是分别共享边和面。Mg2+通常插入到空腔1和2中,而空腔3中共面的Mo原子对Mg2+具有高静电排斥力。
对于MgxMo6S8 (0<xMg<2),Mg在室温下可进行嵌入和脱出。如图3(c)所示,Mg2+有12个可能的位点,6个内部(橙色)和6个外部(蓝色),其中内部-内部(以及外部-外部)位点之间扩散距离相对较短(约0.9 ),而内部-外部位点之间的距离较长(约2.0 )。因此,Mg2+优先在内部位点扩散,内部-外部互扩散的可能性较低[90]。接着,根据第一性原理计算,Mg占据内部位点后,其浓度影响Mo6S8的空间位阻,导致Mg2+在外部位点比内部位点的迁移势垒高出约275 meV[91]。因此,Mg2+在Mo6S8中的迁移数有限且迁移速率缓慢,导致了动力学和容量不足。
如图3(d)所示,AURBACH等[92]使用Mo6S8正极在Mg(AlCl2BuEt)2/THF电解液中匹配Mg负极,两个充放电平台分别代表第1个Mg2+和第2个Mg2+的可逆插层(见式(3)和(4)),可实现122 mA·h/g的比容量,循环2500次后容量几乎不衰减。
Mg2++Mo6S8+2e→MgMo6S8 (3)
Mg2++MgMo6S8+2e→Mg2Mo6S8 (4)
WAN等[93]报道,在氯化物为电解质的THF中,Mg被阴离子Cl-强配位,在溶液中形成单电荷的MgCl+和。在电池放电过程中,将Mg插入Chevrel相需要打破强离子Mg—Cl键。通过对Chevrel相表面物质的吸附能计算,表明了Mo有助于打破Mg—Cl键与Cl-结合,而S原子则促进Mg2+插入体相Mo6S8(见图3(e))。
尽管Chevrel相Mo6S8显示出了极好的Mg插层热力学可逆性,但其有限的比容量和较差动力学限制了其实际应用。更为重要的是亚稳态Mo6S8的合成需要先合成CuMo6S8,然后将Cu原子从Cu2Mo6S8中酸浸出,同时合成条件需要严格控制惰性气氛和压力,这种合成方式耗时耗力不适用于大规模产业化。因此,开发其他硫化物正极用于可充镁电池成为了必然选择。
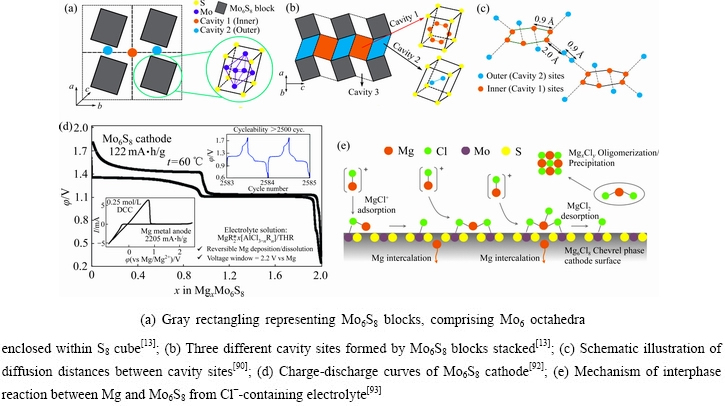
图3 Chevrel相结构;Mo6S8格堆积形成的3种空腔;Mg2+在空腔之间的扩散距离;Mo6S8的充放电曲线;Mo6S8在含Cl-的电解质中与Mg的界面反应机制
Fig. 3 Crystal structures of Cheverl phases
3.2 硫化物
层状结构的MoS2和TiS2可以为离子扩散提供二维平面的扩散通道,但是Mg2+在层间的扩散能垒高而造成Mg2+的缓慢扩散动力学。姚彦课题组[95]使用聚环氧乙烷(PEO)插入MoS2层间,将其层间距6.2 膨胀到14.5 ,计算表明Mg2+的扩散速率提升2个数量级,首次放电比容量从~20 mA·h/g提升至~80 mA·h/g。不过,对MoS2进行层间距的膨胀操作,需要使用正丁基锂对MoS2进行锂化,使其剥离成单层,这种合成过程繁琐且产量不高,对实用化并不友好。
Mg2+在材料体相中的扩散缓慢,另一原因是需要进行Mg—Cl键的断裂,而这个过程需要至少3 eV的高活化能量[95]。姚彦课题组进一步研究发现MgCl+在没有Mg—Cl键的断裂情况下具有快速的扩散动力学[95],他们使用1-丁基-1-甲基吡咯烷离子(PY14+)将TiS2的层间距从5.69 膨胀到10.86 ,促进MgCl+在TiS2层间的快速扩散,在25 ℃下,每个Ti可以相对应可逆地插入一个MgCl+,比容量可达240 mA·h/g。不过,MgCl+插入PY14+修饰的TiS2层间,使其层间距继续膨胀到18.63 ,体积膨胀将近一倍,这会严重降低实用化镁电池器件的体积能量密度。SHEN等[96]使用十六烷基三甲基溴化铵(CTAB)有机小分子插入CuS层间,并将其层间距从8.2 膨胀到13 ,在Mg[B(hfip)4]2/DME电解液中实现了Mg2+在CuS层间的可逆嵌入和脱出。
SUN等[97]报道了Ti2S4尖晶石,在60 ℃和0.2C倍率下,容量接近理论值的80%,230 W·h/kg的比能量则是Chevrel相的两倍。LIU等[98]随后通过DFT计算了Mg2+在Ti2S4中的扩散活化能为600 meV,小于尖晶石结构的MnO2的扩散活化能(>650 meV)。同时计算了Cr2S4和Mn2S4扩散活化能,分别为540 meV和520 meV;说明尖晶石硫合物相对于尖晶石氧化物具有更好的Mg2+迁移率,但这种高迁移率也是以较低的工作电压和理论比能量为代价的。
VS4由V和二硫阴离子[S2]2-形成一维线性链构型,具有较大的链间间距(5.83 ),可实现Mg的可逆嵌脱[99-100]。在Mg嵌入过程中,阳离子V和阴离子S同时发生变价,即V4+氧化为V5+,[S2]2-还原为S2-;使得VS4可容纳的Mg的量大大增加,可实现250 mA·h/g的首次放电比容量[99]。类似地,黄铁矿型Fe0.5Co0.5S2也被证明在Mg2+插入后,过渡金属阳离子和S阴离子都参与了氧化还原反应贡献了容量[101],首次放电也达到250 mA·h/g比容量。
3.3 氧化物
现有的研究已经将硫化物的首次放电比容量稳定在200 mA·h/g以上,但是放电平台在1 V左右,以至于能量密度还不到商业化锂离子电池的三分之一,甚至不如水系锌离子电池[102]。因此,氧化物作为高电压的可充镁电正极材料被赋予众望。
3.3.1 五氧化二钒
早在1994年,镁离子以电化学方式插层五氧化二钒(V2O5)就已被开展研究[103]。V2O5储存Mg的理论能量密度高达~660 W·h/kg,工作电压可在~2.5 V[104]。如图4(a)所示,V2O5晶格由边和角共享的VO5锥体组成的层交替形成,黄色球体代表层与层之间可嵌入的Mg。正交V2O5有α和δ两种晶型,不同点在于沿着a轴方向上(垂直于b-c面)的堆叠层有所变化,其中蓝色虚线框突出显示了正交结构。1987年,PEREIRA-RAMOS等[105]便证实Mg在高温下(150 ℃)可实现电化学插入正交V2O5结构中。2019年,FU等[104]利用XAS、Raman和XPS表征方法,阐明了V2O5的循环可逆性,且MgxV2O5在镁离子插入过程中形成了新的富镁相。
镁离子电化学嵌入正交晶系V2O5的动力学往往不佳,这主要是由于高电荷密度Mg2+在高离子浓度的正交V2O5晶格中的扩散受到阻碍。一种策略便是在正极晶格中提供“溶剂”分子,以静电方式“屏蔽”Mg2+的电荷与晶格离子的排斥力[106]。为了提高Mg在正交V2O5室温下的扩散速率,NOV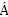 K等[107]试图用水作为共夹层介质来促使Mg2+在V2O5晶格中迁移。当V2O5层间插入水,变成V2O5·H2O干凝胶,V2O5会形成“双层”排列(见图4(b))。V2O5·nH2O是亚稳态,温度加热超过~250 ℃,会脱水转变成正交的V2O5[108],这种结构可以插入Mg2+,Ca2+和Zn2+等离子[109-112]。2018年,RASTGOO-DEYLAMI等[113]首次合成正交晶系的H2V3O8(V3O7×H2O)。在60 ℃下,该材料初始放电容量231 mA·h/g,平均放电电压1.9 V vs Mg/Mg2+,能量密度可达到440 W·h/kg,电化学性能优异。SA等[112]通过NMR、PDF和XANES证实了镁络合物在V2O5·nH2O插入,随着镁化的继续发生,络合物发生分解,并填充在晶格空隙中,结晶水被“挤出”晶格,这可能对镁金属负极产生不利的影响。类似地,通过水的插入将单斜的WO3改变为层状的WO3·H2O也开展了研究[114-115]。
K等[107]试图用水作为共夹层介质来促使Mg2+在V2O5晶格中迁移。当V2O5层间插入水,变成V2O5·H2O干凝胶,V2O5会形成“双层”排列(见图4(b))。V2O5·nH2O是亚稳态,温度加热超过~250 ℃,会脱水转变成正交的V2O5[108],这种结构可以插入Mg2+,Ca2+和Zn2+等离子[109-112]。2018年,RASTGOO-DEYLAMI等[113]首次合成正交晶系的H2V3O8(V3O7×H2O)。在60 ℃下,该材料初始放电容量231 mA·h/g,平均放电电压1.9 V vs Mg/Mg2+,能量密度可达到440 W·h/kg,电化学性能优异。SA等[112]通过NMR、PDF和XANES证实了镁络合物在V2O5·nH2O插入,随着镁化的继续发生,络合物发生分解,并填充在晶格空隙中,结晶水被“挤出”晶格,这可能对镁金属负极产生不利的影响。类似地,通过水的插入将单斜的WO3改变为层状的WO3·H2O也开展了研究[114-115]。
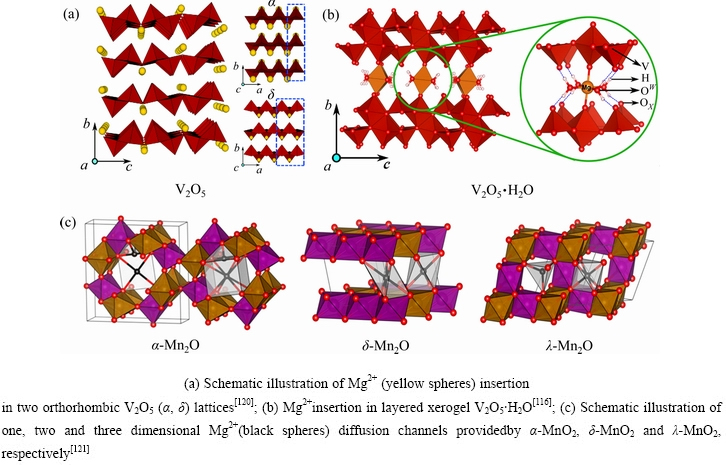
图4 Mg2+在几种氧化物晶格中的插层位点示意图
Fig. 4 Structure of different oxides with Mg intercalation sites
大部分的研究表明,随着电解液中水含量的增加,V2O5放电容量显著增加,但是却与材料中镁含量的变化不匹配[116-117]。因此,需要区分容量提升的原因是镁离子插入或其他电化学反应(如H+插入)。SA等[117]通过固体核磁共振(NMR)方法证实高含水量电解质的高容量主要来源于可逆的质子插入。然而CABANA等[118]研究表明,H2O在ζ-V2O5结构中对Mg2+的插入并非是直接的增强作用,反而促进了插层反应与转化反应的竞争,不利于Mg2+的插入。因此电解液中的H2O对氧化物的作用也不是绝对的。ESPARCIA等[119]直接合成钒铜铵(NH4V4O10),NH4+在V2O5层间起到与H2O一样的作用,促进了Mg2+在V2O5层间的插入,但不会造成电化学循环过程中,晶格水流失进入非水系电解液中发生分解等问题。使用除水分子外的不同的无机或有机小分子插层改性V2O5将是一个不错的尝试。
3.3.2三氧化钼和二氧化钛
层状结构的MoO3作为镁的插层正极在1995年也被研究[122],但镁的嵌入量非常有限[123]。当在MoO3中引入F取代O得到MoO2.8F0.2,并因此改变其与邻近Mo的电子相互作用,使得原有晶格发生畸变,有利于Mg的嵌入脱出[124]。对MoO3进行V掺杂得到Mo2.5+yVO9+δ (Mo5+/6+, V4+/5+),V离子和Mo离子之间的强库伦相互作用互相抵消,有助于缓解镁扩散动力学缓慢的问题,以及Mg2+插入过程中,两种离子的电荷重新分配有助于减少体积膨胀[125]。
锐钛矿型TiO2是锂离子电池和钠离子电池的一种候选大功率电极材料,但直接作为储Mg的正极材料,储Mg量非常有限(MgxTiO2, x=0.1)[126]。在TiO2中引入Ti空位[127]和氧空位[128-129],能够为Mg提供占据位点,实现晶格间大量储Mg,将放电比容量从25 mA·h/g提升至165 mA·h/g。YANG等[130]报道的质子修饰的具有Ti空位的TiO2纳米薄片,质子的存在促进了Cl-的剥离,促使Mg2+具有快速的扩散动力学,常温下放电比容量可达250 mA·h/g。
3.3.3 二氧化锰
二氧化锰具有丰富的晶型结构同时成本低廉和环境相容性好,因此在1991年,BRUCE等[131]便研究了Mg嵌入多种晶型的MnO2。在MnO2中,1个Mn原子与6个O原子配位形成MnO6八面体,这种八面体模块以不同方式堆积会形成多种晶体结构,可分为一维隧道结构、二维层状结构和三维网状结构。一维隧道结构可以细分为2×2模块堆积成隧道的α-MnO2 (碱硬锰矿),1×1模块堆积形成隧道的β-MnO2 (软锰矿)和2×1的γ-MnO2 (六方锰矿);以及2×3模块堆积的钡硬锰矿(romanechite)和3×3的钡镁锰矿(Todorokite)等。二维层状结构以δ-MnO2 (birnessite, 水钠锰矿)为主,三维网状结构以λ-MnO2 (Spinel, 尖晶石)为主。
如图4(c)所示,一维隧道2×2结构的α-MnO2,提供了较大的离子通道(~5 ),远大于Mg2+的半径(0.86 )。ZHANG等[132]通过X射线吸收近边结构(XANES)证实了Mg2+在α-MnO2孔道结构中能够进行可逆地嵌入和脱出,同时扩展X射线吸收精细结构(EXAFS)发现Mg2+在a-MnO2嵌脱过程中存在不可逆的转化反应导致首次放电和充电的容量损失。ARTHUR等[133]进一步研究发现,这种不可逆的转化反应是MgO的生成所致。
NAM等[134]将尖晶石的Mn3O4置于MgSO4水溶液中,电化学循环50次后,发现伴随着Mn2+的溶解和Mn3+的氧化,Mn3O4进而转变为层状的水钠锰矿d-MnO2,期间水分子插入层间减弱Mg2+与阴离子O2-的静电相互作用,从而促使Mg2+更容易在δ-MnO2层间脱嵌。KIM等[135]使用扫描透射电子显微镜(STEM)对尖晶石的Mn3O4在水分子的诱导下向层状δ-MnO2转变进行直接观察。虽然,H2O分子在晶格内部屏蔽了Mg2+与晶格氧阴离子的强库伦力,有效增强了Mg2+扩散能力,但是H2O会在金属镁负极表面形成MgO钝化层,导致过电位大幅增加[107]。
相比于硫化物,氧化物具有电压高,稳定性好,制备方法简单,成本低廉和环境友好等特点。然而,氧化物在Mg2+嵌入时,容易转化生成热力学更为稳定的MgO;但该转化反应是不可逆反应,这严重阻碍了这些氧化物作为镁电正极插层材料的应用。
3.4 高电压插层型正极材料
3.4.1 尖晶石化合物
近年来,尖晶石相的MgM2O4 (M=V, Cr,Mn, Fe, Co, Ni)等被证明可以实现镁的可逆脱嵌[136-137],同时尖晶石氧化物是镁电池突破3.0 V高电压的正极材料[138]。SINHA等[139]首次报道了Mg2+插入尖晶石Mn2O4 (λ-MnO2,见图4(c))中,通过将LiMn2O4置于Mg(NO3)2水溶液中,进行电化学循环。充电时Li脱出后,形成λ-MnO2,然后放电时Mg2+插入形成尖晶石MgMn2O4。近些年的研究直接合成含镁的MgMn2O4[137],但是Mg2+在尖晶石氧化物中的迁移率通常仍然较低,同时Mg2+/Mn2+在尖晶石结构中的倒置,导致了尖晶石相会转变成岩盐相结构[140]。本应在八面体位(16c)的Mn位,会被部分Mg占据;同时,四面体位(8a)的Mg被部分Mn所占据。阳离子倒置会造成尖晶石结构变化而阻碍Mg2+扩散通道[141]。通过DFT计算和NMR表征,MgCr2O4比MgMn2O4具有更低的Mg2+扩散能垒(650 meV vs 800 meV)[142-143]。
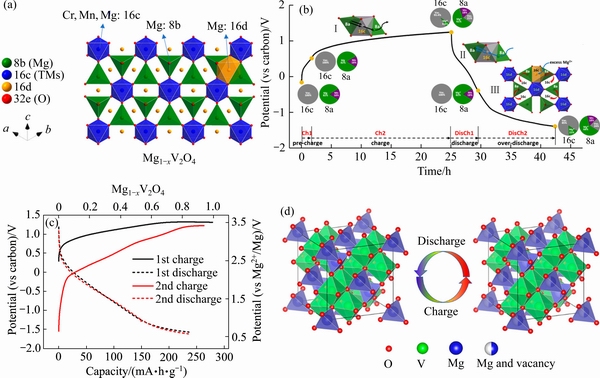
图5 MgCrMnO4尖晶石的结构;MgCrMnO4在第一次充放电过程中Mg2+迁移路径的示意图; MgV2O4纳米晶的充放电曲线;MgV2O4充放电过程中Mg2+的可逆脱出和嵌入的结构示意图
Fig. 5 Structure of MgCrMnO4 spinel[145](a); Schematic illustration of Mg2+ migration processes of MgCrMnO4 during the first charge/discharge[146](b); Charge/discharge profiles of MgV2O4 nanocrystals[147](c); Schematic illustration of Mg2+ reversible de/intercalation in MgV2O4 during charge /discharge[147](d)
MgCr2O4中镁的脱出和嵌入电压在3.7 V vs Mg/Mg2+,但镁电解液中的醚类溶剂的耐受电压仅为3.5 V,同时,高价的氧化态Cr4+在主体晶格中并不稳定[137-138]。对八面体位的Cr进行缺陷调控造Cr空位有利于促进Mg2+的扩散迁移[144]。或着,将Cr与Mn进行混合一并占据八面体位(16c)构筑固溶体MgCrMnO4(见图5(a));Cr3+为Mg2+提供高扩散迁移率,Mn3+为Mg2+提供合适的高氧化还原电位插层反应[145]。YIN等[146]通过高分辨XRD和中子衍射数据研究发现,Cr3+的掺杂还可以有效缓解尖晶石中Mg/Mn晶石倒置率从18%降低至10%,进一步提高了Mg2+迁移率。通过电化学原位XRD观测MgCrMnO4正极中在循环过程中的结构演变,在Mg2+脱出后,尖晶石CrMnO4晶格没有发生相变,表明在3.0 V的高电势下,其结构稳定(见图5(b))。Mg2+的插入伴随着两个均匀的固溶反应,涉及四面体8a和八面体16c位点之间的阳离子重新分布。这些阳离子运动是由充电时8a位点上的空位或过放电时16c位点上过量的Mg2+的插入驱动的。相反,过渡金属和倒置的Mg2+在16d位保持不变。
CABANA课题组[147]证实了MgV2O4纳米晶可以实现高容量的Mg2+脱出,使用MgTFSI2电解质在PY14TFSI离子液体作为的溶剂中,实现3.5 V充放电,在110 ℃下可实现首次放电超过200 mA·h/g(见图5(c))。图5(d)所示,在充电时,Mg2+从尖晶石结构的四面体位脱出,相反,放电时,Mg2+扩散进入四面体空位。同样地,将Cr与V进行混合构筑固溶体MgCrVO4[142],可以利用Cr提升Mg2+迁移率,增加尖晶石与镁的电化学反应活性。与锂离子不同,钴基尖晶石的镁嵌入动力学较差[13]。同时SA等[148]观测到MgCo2O4在脱嵌Mg2+时,均有不可逆的MgO生成。对于尖晶石MgFe2O4结构,Fe3O4与MgO构成的异质结创造了富氧环境促进Mg2+容易掺入Fe2+位点[149],导致Mg1-xFe2+xO4尖晶石结构的形成,其中部分Fe2+被氧化为Fe3+。
尖晶石相的ZnM2O4 (M=Fe, Co)也被证实在Mg的插入后,发生了向岩盐相结构的转变[150]。SHIMOKAWA等[151]设计了一种缺陷型尖晶石ZnMnO3,Zn强烈倾向于四面体环境而使尖晶石型结构稳定,同样抑制了Mg2+迁移中发生的尖晶石相向岩盐相的转变。MgCr2S4[152-153]和MgIn2S4[154]尖晶石硫化物也可作为正极材料。CEDER课题组[155]通过DFT计算,结合NMR谱表征和EIS电化学测量,预测了MgSc2Se尖晶石硫族化合物具有常温下10-4 S/cm的高镁离子迁移率。
3.4.2 层状和聚阴离子氧化物
锂离子电池正极材料使用的层状结构的氧化物,Li+在层间的脱出和嵌入的扩散速率较快[156-157]。INSHIDA等尝试对锂电层状三元结构进行化学脱锂处理[158-160],则在高温下可以插入有限比例的Mg,最大确证容量为116 mA·h/g。然而,对放电状态的结构研究表明,Mg在过渡金属层中的占位,具有不可逆性且会引起容量衰减。因此,Mg2+已经在锂电层状正极材料中扮演了支撑骨架离子,不容易在层间脱嵌,这种直接复制锂电的经验在电化学动力学上实现起来并不简单。王春生课题组[54]通过离子交换法将层状NaMnO2脱钠得到层状Mg0.15MnO2,少量Mg在MnO2层间扮演骨架支撑离子,匹配多齿甲氧乙基胺螯合溶剂改进的Mg(TFSI)2/DME电解液,可在常温下实现3.3 V高电压的充放电循环,Mg2+在Mg0.15MnO2的可逆脱嵌,比容量可达200 mA·h/g。
从锂离子电池的经验来看,以橄榄石结构为主的聚阴离子化合物具有高的工作电压和良好的循环性能[161]。LING等[162]通过DFT计算多种橄榄石结构氧化物,发现FePO4具有高的嵌镁氧化还原电位和较小的晶格体积变化。事实上,早在2003年,POUL等[163]证明在FePO4中可以插入和脱出多达0.38个Mg2+。不过,这是在极缓慢的充放电速率下进行的,在一般或较高充放电速率下,镁插入FePO4会因橄榄石晶体结构的不稳定性,导致这个过程无法进行,无法发挥出FePO4的理论储镁容量[164]。ORIKASA等[165]制备了具有高度缺陷的碳复合的FePO4材料,可以作为Mg2+的可逆嵌入/脱出,在1.5~3.5 V电压窗口下实现了170 mA·h/g的放电比容量。
聚阴离子氧化物如MgFeSiO4[166-1168],MgMnSiO4[169-171]和MgCoSiO4[172]作为可充镁电池正极材料也被相继报道。然而,不论是尖晶石还是聚阴离子化合物中,所存在的镁离子动力学缓慢的问题,都需要通过有效的材料设计来解决。目前来看,材料缺陷化造镁离子和氧空位以及材料纳米化缩短镁离子扩散路径的两种途径较为有效。
3.5 高容量的转化型正极材料
3.5.1 硫正极
镁离子插入材料晶格内部发生的插层反应,受限于热力学上晶格内部储镁位点的有限和动力学上高电荷密度镁离子的扩散缓慢,而且镁插入晶格内部会造成材料晶格的扭曲,长期嵌入脱出循环会发生材料结构坍塌甚至粉化的问题。相比于插层型正极材料,硫(S)作为转化型正极直接与镁发生反应生成硫化镁化合物,具有更高的放电比容量(理论质量比容量1675 mA·h/g和理论体积比容量3459 mA·h/cm3),同时这个反应是可逆进行的[173]。
镁硫电池与锂硫、钠硫等金属/硫电池一样,需要克服中间产物在电解液中溶解和穿梭的问题,这需要从电极材料设计和电解液的筛选入手[174]。此外,硫作为亲电子正极,需要匹配非亲核电解液,以免与电解液直接发生反应。2011年,KIM和MULDOON等[41]第一次报道了基于MgCl2(HMDS)-AlCl3非亲核电解液的镁硫电池,1200 mA·h/g和2484 mA·h/cm3的高比容量的特性吸引了不少研究人员的注意。GAO等[175]通过X射线光电子能谱(XPS)分析认为镁硫电池放电路径与锂硫电池类似,为S8→MgS8→MgS2→MgS3多个过程,多硫化物与Mg负极表面的副反应是电池失效的原因之一。
硫正极与金属镁负极构成的镁硫电池在低电压下具有高容量和可逆电化学活性,但涉及到镁硫电池的一些基础科学问题还在研究中,以及涉及的工艺技术难题还没有得到有效的解决。充放电过程中正极的严重过充和硫的低利用率,镁多硫化物的形成和镁离子扩散存在缓慢的动力学,以及与锂硫电池一样存在的硫化物的“穿梭”效应等“疑难杂症”,这些问题还需从正极固硫、镁负极保护和亲核电解液设计等继续开展研究。
3.5.2 氧和碘正极
氧作为正极和金属镁负极可以构成镁空气电池,理论工作电压为3.1 V,能量密度高达6800 W·h/kg[176]。2013年,SHIGA等[177]提出了一种利用碘-二甲基亚砜(I2-DMSO)复合物实现可充镁空气电池的充放电循环。将I2加入到溶剂DMSO中,可以使MgO发生溶解,有利于Mg和O2的可逆反应。在1.25 V (vs Mg/Mg2+)的放电电压平台下,具有1000 mA·h/g的放电比容量。不过,这种电解液无法与镁负极兼容,不可以做到镁的可逆沉积。2016年,王春生课题组避开了Mg和O2的反应,直接使用多孔碳/碘复合正极与镁发生液固反应[178],其中可溶的I2与Mg2+反应生成可溶的中间体,然后形成不溶的最终产物MgI2。
镁空气电池与锂空气电池所遇到的难题相近,中间产物过氧化镁的可逆反应和正极/电解液界面的稳定均是需要解决的难题,因而,镁空气电池的实用化才刚刚开始,除了技术瓶颈以外,还有很多基础科学问题亟待解决。
3.5.3 有机分子正极
高电荷密度的Mg2+在无机材料紧密排列晶格中迁移较为困难,而有机材料作为正极,比表面积大且空间分布广,可以使得Mg2+从电解液到电极材料上的迁移途径缩短,同时受到的分子间作用力更小。此外,有机材料的柔韧特性,特别适用于下一代的能源存储场景,如柔性和可穿戴电子产品。此外,有机材料的组成以C和O元素为主,不使用有毒的重金属,这便是低成本、可持续、可回收和安全性高的电极材料[179]。
有机正极材料多以醌或其衍生物为主[180],醌上的双键氧参与电荷反应,苯环作为骨架材料稳定电极结构。相比于有机小分子,有机高分子的聚合物电极在稳定电极结构方面更有优势。2018年,姚彦等[181]使用醌基聚合物正极在Mg(CB11H12)2电解液中,实现了Mg2+的直接存储,能量密度和功率密度最高可达243 W·h/kg和3.4 kW/kg,循环2500次容量依然保持为87%。2020年,他们进一步设计了1,2-苯醌衍生物芘-4,5,9,10-四酮(PTO)[62],不同于镁离子在电极中的固相反应,该反应利用了氧化还原烯醇化机理,PTO还原为可溶的Mg1PTO中间态,接着还原为不溶的Mg2PTO。此反应巧妙地绕过了镁离子固相扩散缓慢的问题,避免了转化型材料动力学缓慢的问题,最终实现了313 W·h/kg的能量密度和30.4 kW/kg的超高功率密度。
3.6 高功率活性炭正极材料
超高比表面积的活性炭是商业化双电层超级电容器的电极材料[182-184]。AURBACH等[34]将活性炭作为正极,匹配金属镁负极组成了混合型超级电容器。活性炭/镁基混合型电容器分别在APC-LiCl电解液中,在2.4 V的工作电压下,实现了90 F/g的比电容和数千次循环。随后,CABANA等[185]对活性炭匹配金属镁负极的工作机制进行了详细阐述,这种混合型电容器如锂离子电池一样,遵循“摇椅式”机制。充电时,镁离子在活性炭表面脱附,对应的镁离子在金属镁负极沉积;相反,当放电时,镁离子在活性炭表面吸附,对应的镁离子在金属镁负极溶解。接着他们使用Mg(TFSI)2电解液用于混合型电容器,实现了104.5 F/g的高比电容。
活性炭正极匹配金属镁负极组成的混合型电容器,不仅利用了镁的高比容量,也避开了高电荷密度的Mg2+在其他正极中扩散困难的问题;其次这种电容器的能量密度高于传统的双电层超级电容器,并缓解了电容器存在的自放电问题。
3.7 小结
从可充镁正极材料发展来看,借鉴锂离子电池正极材料的成功经验是可取的,但是需要明晰的是,二价的Mg2+不论是在电极材料内部还是电极/电解液界面,都是具有与Li+不同的电化学反应机制[186]。
高电压的尖晶石氧化物的可逆镁插层和高容量的硫正极的可逆镁转化反应代表着镁电正极材料的两种发展路径,两种正极材料的改性策略也因插层和转化反应机制不同而有较大区别。高电荷密度的Mg2+在高电压的尖晶石氧化物中扩散迁移缓慢,可以通过缺陷工程制造晶格镁和氧空位,以减弱Mg2+和阴离子晶格氧之间的强静电相互作用;或者对材料进行纳米化缩短Mg2+扩散路径以提高Mg2+扩散速度。高容量的硫正极与镁生成的中间产物多硫化镁的“穿梭”效应可使用多孔碳材料或多孔硫化物等支撑骨架来起到抑制作用,以及适量的催化剂的加入有助于提升镁离子和硫正极的反应动力学。
就目前关于镁电池正极材料的研究来看,正极材料的选择往往还依赖于电池本身的特性。高电压的插层正极材料发生的是镁可逆脱出和嵌入的插层反应,而金属镁负极发生的是镁可逆沉积和溶解反应;在高电压电解液的选择上,不仅要考虑匹配高电压尖晶石或层状氧化物,还要匹配是否可以在金属镁表面发生可逆沉积和溶解反应,这限制了可充镁电池的材料选择和设计。将可充镁电池负极扩展到不仅仅是镁可逆沉积和溶解,还可以实现镁的插层或合金化反应等等。接下来会对此进行介绍和讨论。
4 可充镁电池负极反应机制
4.1 金属镁表面的可逆沉积/溶解
金属锂直接作为锂电池负极,会使得电池中的锂离子在锂负极表面以树枝状沉积(见图6(a)),这直接造成了锂电池严重的安全问题(第2节着重介绍)。然而,使用金属负极可以提供超高的能量密度,因此当人们争相寻找无枝晶的金属负极时,金属镁脱颖而出。如图6(b)所示,镁离子在金属镁负极表面沉积时是以致密状堆积[23]。不过,与金属锂表面形成的固体电解质界面(SEI)膜不同,金属镁负极的表面会因电解液分解等形成氧化镁等组成钝化膜,这阻碍了电极反应的进一步发生[29]。通过在金属镁表面构筑保护层,可以抑制抑制钝化膜的生成。比如,在金属镁表面构筑Ge保护层[187]和三维骨架的Mg3Bi2保护层[188]。
MANDAI等[189]发现镁在单晶镁上的沉积/溶解具有取向性,通常沿着(0001)或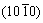 晶面。对镁进行合金化,可以通过调控镁晶粒晶界来提高镁的电化学活性,所制备的镁钙合金具有比纯镁作为镁电负极更小的过电位以及更高的响应电流。
晶面。对镁进行合金化,可以通过调控镁晶粒晶界来提高镁的电化学活性,所制备的镁钙合金具有比纯镁作为镁电负极更小的过电位以及更高的响应电流。
4.2 镁的合金化反应
不同于Mg2+在金属镁表面的可逆沉积/溶解,Mg2+还可以插入Bi[190]、Sb[190]、Sn[191]和In[192]等金属间,发生可逆的合金化反应。2012年,ARTHUR等[190]证明了Bi和Sb可以作为可充镁电池负极。然后,SINGH等[191]将Sn作为负极,当Mg2+插入后生成Mg2Sn合金,首次放电比容量高达900 mA·h/g。金属间化合物BiSb[190]和InBi[193]也可以与Mg2+发生合金化反应,展现出高比容量(>400 mA·h/g)。
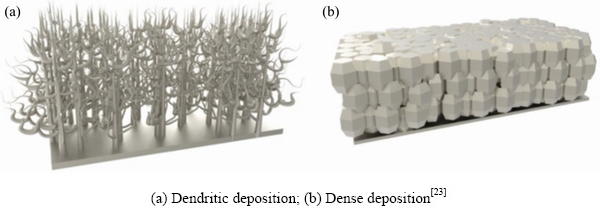
图6 金属离子在金属负极表面沉积的形貌示意图
Fig. 6 Morphology diagrams of metal ion deposited on metal anodes
Mg2+插入金属负极内部,发生的可逆合金化反应提供了高比容量,但是Mg2+插入后金属负极产生的较大体积变化以及插入过程中缓慢的动力学,会造成对负极不可逆的破坏。合成Mg2Sn[194]和Mg3Bi[195-196]等合金直接作为负极可以缓解这种问题。还有一种方法是将金属纳米化,比如将Bi金属做成纳米管状,这种纳米结构可以有效地适应大体积变化,并显著缩短Mg2+的扩散长度[197]。
此外,Si已经成为锂离子电池的商业化负极材料,具有非常高的比容量(生成Li22Si5)。人们试图将硅作为镁离子电池的负极材料,以Mg2Si储存镁的理论比容量为3816 mA·h/g。然而,镁在纳米硅中的可逆电化学存储尚未得到实验证明,尽管理论研究预测硅与镁的合金化反应在热力学上是有利的。基于目前的研究现状,可以电化学充电将镁从Mg2Si中去除,但不能电化学放电将镁引入特征尺寸为100 nm的无氧化纳米结构硅中[198]。
4.3 镁的插层反应
石墨负极已经广泛用于商业化锂离子电池,相比于Li+容易插入石墨层间,高电荷密度的Mg2+插入石墨层难度较大。GOD等[199]研究少量的Mg2+可以插入石墨层间,但是大量的Mg会在石墨表面沉积,同时电解液在石墨表面被还原生成钝化膜。KIM等[200]发现Mg(TFSI)2在乙二醇二甲醚和二乙二醇二甲醚的混合溶剂中,会发生Mg2+和二乙二醇二甲醚溶剂分子共嵌入石墨的反应。尽管在2 mA/g电流下,电池的比容量为180 mA·h/g,但是嵌入和脱出反应过程中有2 V的过电位。更严重的是,溶剂插层会造成石墨本身不可逆的脱落[199, 201]。镁离子不易插入晶格紧密堆积的石墨层间,将石墨剥离得到二维材料石墨烯,有结果表明在含25%缺陷的石墨烯中,Mg2+在大比表面积的缺陷石墨烯层发生吸脱附,获得高达1042 mA·h/g的储镁容量[202]。
WU等[203]报道了镁在钛酸锂(Li4Ti5O12, LTO)中稳定和可逆的电化学插层。在平均电压为0.5 V vs Mg/Mg2+的条件下,当LTO晶体尺寸降低到10 nm以下,其理论容量接近175 mA·h/g。在Mg(AlCl2BuEt)2/THF电解液中,以1C进行了500多次循环后,仍具有95%的容量保持率。随后他们又在电解液中加入LiCl使得镁和锂共插层Li4Ti5O12从而提升了放电比容量[204]。
4.4 小结
可充镁电池直接使用金属镁做负极的初衷是利用其高能量密度,但是与活性过高的金属锂恰恰相反,镁的惰性导致其可逆溶解/沉积的库伦效率无法达到百分之百,而且在匹配镁电解液和正极材料的选择上也有很大的限制。例如,具有超过4 V高电压稳定性的碳酸酯类溶剂与金属镁会发生钝化反应,但是通过在镁负极表面构筑一层人工电解质界面膜,可以使Mg2+通过这层界面膜实现在碳酸酯溶剂中的可逆镁沉积和溶解[205]。因此,在匹配高电压氧化物和高电压电解液时,需要对金属镁进行改性或者还可考虑使用插层或转化材料等作负极;在匹配高容量的硫和硫化物复合正极时,使用镁沉积库伦效率更高的镁合金则往往是更好的选择。
5 结论与展望
目前,可充镁电池还未被商业化,尽管可充镁电池同锂电池一同在20世纪70年代石油危机大背景下便开始被研究。锂离子电池的成功是在基础科学和工艺技术工作者们的相互促进和扶持下,伴随着插层材料的发现以及后续人力物力的投入和不断改进工艺技术所产生的巨大进步。虽然锂离子电池的成功经验可以借鉴,但是二价态的镁离子作为储能介质需要两个电子的转移,其存在的基础科学问题和工艺技术难题与一价态锂离子有诸多不同。
本文从镁电池的发展历史出发,详细地介绍了可充镁电池器件中三种关键组成部分,即电解液、正极和负极材料的研究进展。从目前的研究来看,遵循弱配位的阴离子设计的电解液有助于提高镁离子在电解液和电极/电解液界面的迁移速率;正极材料的缺陷化和纳米化有助于高电荷密度二价镁离子进入材料晶格,并加快镁离子在晶格内部的扩散速率;金属镁负极的表面改性和合金化有助于提升镁离子的可逆沉积和溶解效率且避免过多界面副反应的发生。
可充镁电池未来的应用场景更多的将是在分布式或规模化的储能电站。因此,镁电池的充电和放电的能量转化利用率是非常关键的指标。具体到镁电池的电化学反应来看,由于较高的过电位会造成法拉第效率过低,部分电能在充电和放电过程中还会以热能的形式所损耗[14]。因此,降低电化学反应过程中的过电位将是可充镁电池后续从材料和器件设计方面不断改进的一个重要方向。
在探索可充镁电池商业化应用途径过程中,不仅需要逐步明晰其中所涉及的电化学反应机制、材料和镁离子的相互作用机理等科学问题;也需要不断克服在材料设计和合成以及器件匹配和组装中所遇到的技术有限的瓶颈问题。镁离子与电极材料在发生电化学反应过程中存在的晶格畸变和电荷密度改变等问题,可以利用一些先进光学表征结合原位测试方法,以及密度泛函等材料计算方法来研究和判定。具有纳米尺度和空位缺陷的镁电池正极材料的可控合成,新型弱配位阴离子的电解液的设计合成,以及金属镁负极的表面修饰改性,这些均需要借助先进智能制造技术来实现进一步的低成本和批量化生产。
在全球环境治理和应对气候变化的大背景下,随着加快能源结构转型的“双碳”目标的提出,可充镁电池的实用化研究之路充满了机遇,但也布满了荆棘。不论是对于科学研究者们还是工程技术人员们,这都将是一个值得为之努力的综合性、系统性的大问题。
REFERENCES
[1] 习近平. 共同构建人与自然生命共同体[N]. 人民日报, 2021-04-23(2).
XI Jin-ping. For Man and Nature: Building a Community of Life Together[N]. People’s Daily, 2021-04-23(2).
[2] 黄 震, 谢晓敏. 碳中和愿景下的能源变革[J]. 中国科学院院刊, 2021, 36(9): 1010-1018.
HUANG Zhen, XIE Xiao-min. Energy revolution under vision of carbon neutrality[J]. Bulletin of Chinese Academy of Sciences, 2021, 36(9): 1010-1018.
[3] 梁叔全, 程一兵, 方国赵, 等. 能源光电转换与大规模储能二次电池关键材料的研究进展[J]. 中国有色金属学报, 2019, 29(9): 2064-2114.
LIANG Shu-quan, CHENG Yi-bing, FANG Guo-zhao, et al. Research progress of key materials for energy Photoelectric Conversion and large-scale energy storage secondary batteries[J]. The Chinese Journal of Nonferrous Metals, 2019, 29(9): 2064-2114.
[4] DUNN B, KAMATH H, TARASCON J M. Electrical energy storage for the grid: A battery of choices[J]. Science, 2011, 334(6058): 928-935.
[5] ZHU Z L, LIU H, CHEN J S Y, et al. Electrochemical behavior and electrolytic preparation of lead in eutectic NaCl-KCl melts[J]. Transactions of Nonferrous Metals Society of China, 2020, 30(9): 2568-2576.
[6] 雷舒雅, 徐 睿, 孙 伟, 等. 废旧锂离子电池回收利用[J]. 中国有色金属学报, 2021, 31(11): 3303-3319.
LEI Shu-ya, XU Rui, SUN Wei, XU Sheng-ming, YANG Yue, et al. Recycling of spent lithium-ion battery[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(11): 3303-3319.
[7] 李 钊, 王 忠, 班丽卿, 等. 富锂锰基正极材料的表面改性研究进展[J]. 化学学报, 2019, 77(11): 1115-1128.
LI Zhao, WANG Zhong, BAN Li-qin, et al. Recent advances on surface modification of Li- and Mn-rich cathode materials[J]. Acta Chimica Sinica, 2019, 77(11): 1115-1128.
[8] 胡国琛, 胡年香, 伍继君, 等. 锂离子电池正极材料中有价金属回收研究进展[J]. 中国有色金属学报, 2021. doi: 10.11817/j.ysxb.1004.0609.2021-42052.
HU Guo-chen, HU Nian-xiang, WU Ji-jun, et al. Research progress in the recovery of valuable metals in cathode materials for lithium-ion batteries[J]. The Chinese Journal of Nonferrous Metals, 2021. doi: 10.11817/j.ysxb.1004.0609. 2021-42052.
[9] ARMAND M, TARASCON J M. Building better batteries[J]. Nature, 2008, 451(7179): 652-657.
[10] 董 强, 徐海嵩, 潘 飞, 等. 采用溶剂热法制备的钠离子电池用石墨烯基锡铜负极材料[J]. 中国有色金属学报, 2021, 31(4): 931-937.
DONG Qiang, XU Hai-song, PAN Fei, et al. Graphene- based SnCu anode synthesized via solverthermal method for sodium ion batteries[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(4): 931-937.
[11] QIN M L, YIN C Y, XU W, et al. Facile synthesis of high capacity P2-type Na2/3Fe1/2Mn1/2O2 cathode material for sodium-ion batteries[J]. Transactions of Nonferrous Metals Society of China, 2021, 31(7): 2074-2080.
[12] 张 行, 朱黎霞, 王效聪, 等. 二次锌-空气电池锌阳极研究新进展[J]. 中国有色金属学报, 2020, 30(8): 1895-1905.
ZHANG Xing, ZHU Li-xia, WANG Xiao-cong, et al. Latest research progress in zinc anode of secondary Zn-air batteries[J]. The Chinese Journal of Nonferrous Metals, 2020, 30(8): 1895-1905.
[13] CANEPA P, SAI GAUTAM G, HANNAH D C, et al. Odyssey of multivalent cathode materials: Open questions and future challenges[J]. Chemical Reviews, 2017, 117(5): 4287-4341.
[14] JOHNSON I D, INGRAM B J, CABANA J. The quest for functional oxide cathodes for magnesium batteries: A critical perspective[J]. ACS Energy Letters, 2021, 6(5): 1892-1900.
[15] BLAKE I C. Fiftieth anniversary: The anniversary issue on primary cell[J]. Journal of The Electrochemical Society, 1952, 99(8): 202C-203C.
[16] GREGORY T D, HOFFMAN R J, WINTERTON R C. Nonaqueous electrochemistry of magnesium: applications to energy storage[J]. Journal of The Electrochemical Society, 1990, 137(3): 775-780.
[17] AURBACH D, LU Z, SCHECHTER A, et al. Prototype systems for rechargeable magnesium batteries[J]. Nature, 2000, 407(6805): 724-727.
[18] LIN D, LIU Y, CUI Y. Reviving the lithium metal anode for high-energy batteries[J]. Nature Nanotechnology, 2017, 12(3): 194-206.
[19] SEH Z W, SUN J, SUN Y, et al. A highly reversible room-temperature sodium metal anode[J]. ACS Central Science, 2015, 1(8): 449-455.
[20] 傅献彩, 沈文霞, 姚天扬, 等. 物理化学[M]. 5版. 北京: 高等教育出版社, 2006.
FU Xian-cai, SHEN Wen-xia, YAO Tian-yang, et al. Phyical chemistry[M]. 5th ed. Beijing: Higher Education Press, 2006.
[21] 索鎏敏, 李 泓. 锂离子电池过往与未来[J]. 物理, 2020, 49(1): 17-23.
SUO Liu-min, LI Hong. The past, present and future of lithium ion batteries[J]. Physics, 2020, 49(1): 17-23.
[22] YOSHINO A. The birth of the lithium-ion battery[J]. Angewandte Chemie International Edition,, 2012, 51(24): 5798-5800.
[23] LIANG Y, DONG H, AURBACH D, et al. Current status and future directions of multivalent metal-ion batteries[J]. Nature Energy, 2020, 5(9): 646-656.
[24] LU Z, SCHECHTER A, MOSHKOVICH M, et al. On the electrochemical behavior of magnesium electrodes in polar aprotic electrolyte solutions[J]. Journal of Electroanalytical Chemistry, 1999, 466(2): 203-217.
[25] BUCUR C B. Challenges of a rechargeable magnesium battery: A guide to the viability of this post lithium-ion battery[M]. Cham: Springer International Publishing, 2018.
[26] SEYFERTH D. The Grignard reagents[J]. Organometallics, 2009, 28(6): 1598-1605.
[27] EVANS W V, LEE F H, LEE C H. The decomposition voltage of Grignard reagents in ether solution[J]. Journal of the American Chemical Society, 1935, 57(3): 489-490.
[28] EVANS W V, PEARSON R. The ionic nature of the Grignard reagent[J]. Journal of the American Chemical Society, 1942, 64(12): 2865-2871.
[29] BUCUR C B, GREGORY T, OLIVER A G, et al. Confession of a magnesium battery[J]. The Journal of Physical Chemistry Letters, 2015, 6(18): 3578-3591.
[30] POUR N, GOFER Y, MAJOR D T, et al. Structural analysis of electrolyte solutions for rechargeable Mg batteries by stereoscopic means and DFT calculations[J]. Journal of the American Chemical Society, 2011, 133(16): 6270-6278.
[31] AURBACH D, GIZBAR H, SCHECHTER A, et al. Electrolyte solutions for rechargeable magnesium batteries based on organomagnesium chloroaluminate complexes[J]. Journal of The Electrochemical Society, 2002, 149(2): A115.
[32] GOFER Y, CHUSID O, GIZBAR H, et al. Improved electrolyte solutions for rechargeable magnesium batteries[J]. Electrochemical and Solid-State Letters, 2006, 9(5): A257.
[33] MIZRAHI O, AMIR N, POLLAK E, et al. Electrolyte solutions with a wide electrochemical window for rechargeable magnesium batteries[J]. Journal of the Electrochemical Society, 2008, 155(2): A103.
[34] YOO H D, SHTERENBERG I, GOFER Y, et al. A magnesium-activated carbon hybrid capacitor[J]. Journal of The Electrochemical Society, 2014, 161(3): A410-A415.
[35] MULDOON J, BUCUR C B, OLIVER A G, et al. Electrolyte roadblocks to a magnesium rechargeable battery[J]. Energy & Environmental Science, 2012, 5(3): 5941-5950.
[36] MULDOON J, BUCUR C B, OLIVER A G, et al. Corrosion of magnesium electrolytes: Chlorides–the culprit[J]. Energy & Environmental Science, 2013, 6(2): 482-487.
[37] NELSON E G, BRODY S I, KAMPF J W, et al. A magnesium tetraphenylaluminate battery electrolyte exhibits a wide electrochemical potential window and reduces stainless steel corrosion[J]. Journal of Materials Chemistry A, 2014, 2(43): 18194-18198.
[38] DOE R E, HAN R, HWANG J, et al. Novel, electrolyte solutions comprising fully inorganic salts with high anodic stability for rechargeable magnesium batteries[J]. Chemical Communications, 2014, 50(2): 243-245.
[39] LIU T, SHAO Y, LI G, et al. A facile approach using MgCl2 to formulate high performance Mg2+ electrolytes for rechargeable Mg batteries[J]. Journal of Materials Chemistry A, 2014, 2(10): 3430-3438.
[40] SEE K A, CHAPMAN K W, ZHU L, et al. The interplay of Al and Mg speciation in advanced Mg battery electrolyte solutions[J]. Journal of the American Chemical Society, 2016, 138(1): 328-337.
[41] KIM H S, ARTHUR T S, ALLRED G D, et al. Structure and compatibility of a magnesium electrolyte with a sulphur cathode[J]. Nature Communications, 2011, 2: 427.
[42] KIM S S, BEVILACQUA S C, SEE K A. Conditioning-free Mg electrolyte by the minor addition of Mg(HMDS)2[J]. ACS Applied Materials & Interfaces, 2020, 12(5): 5226-5233.
[43] KIM S S, SEE K A. Activating magnesium electrolytes through chemical generation of free chloride and removal of trace water[J]. ACS Applied Materials & Interfaces, 2021, 13(1): 671-680.
[44] LOSSIUS L P, EMMENEGGER F. Plating of magnesium from organic solvents[J]. Electrochimica Acta, 1996, 41(3): 445-447.
[45] HA S Y, LEE Y W, WOO S W, et al. Magnesium(Ⅱ) bis(trifluoromethane sulfonyl) imide-based electrolytes with wide electrochemical windows for rechargeable magnesium batteries[J]. ACS Applied Materials & Interfaces, 2014, 6(6): 4063-4073.
[46] SALAMA M, SHTERENBERG I, GIZBAR H, et al. Unique behavior of dimethoxyethane (DME)/Mg(N(SO2CF3)2)2 solutions[J]. The Journal of Physical Chemistry C, 2016, 120(35): 19586-19594.
[47] RAJPUT N N, QU X, SA N, et al. The coupling between stability and ion pair formation in magnesium electrolytes from first-principles quantum mechanics and classical molecular dynamics[J]. Journal of the American Chemical Society, 2015, 137(9): 3411-3420.
[48] SHTERENBERG I, SALAMA M, YOO H D, et al. Evaluation of (CF3SO2)2N-(TFSI) based electrolyte solutions for Mg batteries[J]. Journal of The Electrochemical Society, 2015, 162(13): A7118-A7128.
[49] YANG L, YANG C, CHEN Y, et al. Hybrid MgCl2/ AlCl3/Mg(TFSI)2 electrolytes in DME enabling high-rate rechargeable Mg batteries[J]. ACS Applied Materials & Interfaces, 2021, 13(26): 30712-30721.
[50] YOO H D, HAN S D, BOLOTIN I L, et al. Degradation mechanisms of magnesium metal anodes in electrolytes based on (CF3SO2)2N– at high current densities[J]. Langmuir, 2017, 33(37): 9398-9406.
[51] SHIMOKAWA K, MATSUMOTO H, ICHITSUBO T. Solvation-structure modification by concentrating Mg(TFSA)2-MgCl2-triglyme ternary electrolyte[J]. The Journal of Physical Chemistry Letters, 2018, 9(16): 4732-4737.
[52] MANDAI T, TATESAKA K, SOH K, et al. Modifications in coordination structure of Mg[TFSA]2-based supporting salts for high-voltage magnesium rechargeable batteries[J]. Physical Chemistry Chemical Physics, 2019, 21(23): 12100-12111.
[53] FAN S, ASSELIN G M, PAN B, et al. A simple halogen-free magnesium electrolyte for reversible magnesium deposition through cosolvent assistance[J]. ACS Applied Materials & Interfaces, 2020, 12(9): 10252-10260.
[54] HOU S, JI X, GASKELL K, et al. Solvation sheath reorganization enables divalent metal batteries with fast interfacial charge transfer kinetics[J]. Science, 2021, 374(6564): 172-178.
[55] CONNOR J H, REID W E, WOOD G B. Electrodeposition of metals from organic solutions[J]. Journal of The Electrochemical Society, 1957, 104(1): 38.
[56] BRENNER A. Note on the electrodeposition of magnesium from an organic solution of a magnesium-boron complex[J]. Journal of the Electrochemical Society, 1971, 118(1): 99.
[57] MOHTADI R, MATSUI M, ARTHUR T S, et al. Magnesium borohydride: From hydrogen storage to magnesium battery[J]. Angewandte Chemie International Edition, 2012, 51(39): 9780-9783.
[58] CARTER T J, MOHTADI R, ARTHUR T S, et al. Boron clusters as highly stable magnesium-battery electrolytes[J]. Angewandte Chemie International Edition, 2014, 53(12): 3173-3177.
[59] TUTUSAUS O, MOHTADI R, ARTHUR T S, et al. An efficient halogen-free electrolyte for use in rechargeable magnesium batteries[J]. Angewandte Chemie International Edition, 2015, 127(27): 8011-8015.
[60] JAY R, TOMICH A W, ZHANG J, et al. Comparative study of Mg(CB11H12)2 and Mg(TFSI)2 at the magnesium/ electrolyte interface[J]. ACS Applied Materials & Interfaces, 2019, 11(12): 11414-11420.
[61] HAHN N T, SEGUIN T J, LAU K C, et al. Enhanced stability of the carba-closo-dodecaborate anion for high- voltage battery electrolytes through rational design[J]. Journal of the American Chemical Society, 2018, 140(35): 11076-11084.
[62] DONG H, TUTUSAUS O, LIANG Y, et al. High-power Mg batteries enabled by heterogeneous enolization redox chemistry and weakly coordinating electrolytes[J]. Nature Energy, 2020, 5(12): 1043-1050.
[63] GEIGER W E, BARRIERE F. Organometallic electrochemistry based on electrolytes containing weakly- coordinating fluoroarylborate anions[J]. Accounts of Chemical Research, 2010, 43(7): 1030-1039.
[64] LUO J, BI Y, ZHANG L, et al. A stable, non-corrosive perfluorinated pinacolatoborate mg electrolyte for rechargeable Mg batteries[J]. Angewandte Chemie International Edition, 2019, 58(21): 6967-6971.
[65] REN W, WU D, NULI Y, et al. An efficient bulky Mg[B(Otfe)4]2 electrolyte and its derivatively general design strategy for rechargeable magnesium batteries[J]. ACS Energy Letters, 2021, 6(9): 3212-3220.
[66] LI Z, DIEMANT T, MENG Z, et al. Establishing a stable anode-electrolyte interface in mg batteries by electrolyte additive[J]. ACS Applied Materials & Interfaces, 2021, 13(28): 33123-33132.
[67] WANG H, FENG X, CHEN Y, et al. Reversible electrochemical interface of Mg metal and conventional electrolyte enabled by intermediate adsorption[J]. ACS Energy Letters, 2020, 5(1): 200-206.
[68] KEYZER E N, GLASS H F J, LIU Z, et al. Mg(PF6)2-based electrolyte systems: understanding electrolyte-electrode interactions for the development of Mg-ion batteries[J]. Journal of the American Chemical Society, 2016, 138(28): 8682-8685.
[69] HERB J T, NIST-LUND C A, ARNOLD C B. A fluorinated alkoxyaluminate electrolyte for magnesium-ion batteries[J]. ACS Energy Letters, 2016, 1(6): 1227-1232.
[70] LAU K C, SEGUIN T J, CARINO E V, et al. Widening electrochemical window of Mg salt by weakly coordinating perfluoroalkoxyaluminate anion for Mg battery electrolyte[J]. Journal of The Electrochemical Society, 2019, 166(8): A1510-A1519.
[71] ZHAO-KARGER Z, GIL BARDAJI M E, FUHR O, et al. A new class of non-corrosive, highly efficient electrolytes for rechargeable magnesium batteries[J]. Journal of Materials Chemistry A, 2017, 5(22): 10815-10820.
[72] SCHWARZ R, PEJIC M, FISCHER P, et al. Magnesocene- based electrolytes: A new class of electrolytes for magnesium batteries[J]. Angewandte Chemie International Edition, 2016, 55(48): 14958-14962.
[73] GIFFIN G A. Ionic liquid-based electrolytes for “beyond lithium” battery technologies[J]. Journal of Materials Chemistry A, 2016, 4(35): 13378-13389.
[74] NULI Y, YANG J, WU R. Reversible deposition and dissolution of magnesium from BMIMBF4 ionic liquid[J]. Electrochemistry Communications, 2005, 7(11): 1105-1110.
[75] WANG P, NULI Y, YANG J, et al. Mixed ionic liquids as electrolyte for reversible deposition and dissolution of magnesium[J]. Surface and Coatings Technology, 2006, 201(6): 3783-3787.
[76] YOSHIMOTO N, MATSUMOTO M, EGASHIA M, et al. Mixed electrolyte consisting of ethylmagnesiumbromide with ionic liquid for rechargeable magnesium electrode[J]. Journal of Power Sources, 2010, 195(7): 2096-2098.
[77] IKEDA S, TAKAHASHI M, ISHIKAWA J, et al. Solid electrolytes with multivalent cation conduction. 1. Conducting species in MgZrPO4 system[J]. Solid State Ionics, 1987, 23(1/2): 125-129.
[78] IMANAKA N, OKAZAKI Y, ADACHI G Y. Divalent magnesium ion conducting characteristics in phosphate based solid electrolyte composites[J]. Journal of Materials Chemistry, 2000, 10(6): 1431-1435.
[79] IMANAKA N, OKAZAKI Y, ADACHI G Y. Divalent magnesium ionic conduction in the magnesium phosphate based composites[J]. Chemistry Letters, 1999, 28(9): 939-940.
[80] PARK B, SCHAEFER J L. Review—polymer electrolytes for magnesium batteries: Forging away from analogs of lithium polymer electrolytes and towards the rechargeable magnesium metal polymer battery[J]. Journal of the Electrochemical Society, 2020, 167(7): 070545.
[81] CHUSID O, GOFER Y, GIZBAR H, et al. Solid-state rechargeable magnesium batteries[J]. Advanced Materials, 2003, 15(7/8): 627-630.
[82] DU A, ZHANG H, ZHANG Z, et al. A crosslinked polytetrahydrofuran-borate-based polymer electrolyte enabling wide-working-temperature-range rechargeable magnesium batteries[J]. Advanced Materials, 2019, 31(11): 1805930.
[83] AUBREY M L, AMELOOT R, WIERS B M, et al. Metal- organic frameworks as solid magnesium electrolytes[J]. Energy & Environmental Science, 2014, 7(2): 667-671.
[84] MINER E M, PARK S S, DINCA M. High Li+ and Mg2+ conductivity in a Cu-azolate metal-organic framework[J]. Journal of the American Chemical Society, 2019, 141(10): 4422-4427.
[85] HEERE M, HANSEN A L, PAYANDEH S, et al. Dynamics of porous and amorphous magnesium borohydride to understand solid state Mg-ion-conductors[J]. Scientific Reports, 2020, 10(1): 9080.
[86] YAN Y, DONONELLI W, JORGENSEN M, et al. The mechanism of Mg2+ conduction in ammine magnesium borohydride promoted by a neutral molecule[J]. Physical Chemistry Chemical Physics, 2020, 22(17): 9204-9209.
[87] LE RUYET R, FLEUTOT B, BERTHELOT R, et al. Mg3(BH4)4(NH2)2 as inorganic solid electrolyte with high Mg2+ ionic conductivity[J]. ACS Applied Energy Materials, 2020, 3(7): 6093-6097.
[88] SAHA P, JAMPANI P H, DATTA M K, et al. A convenient approach to Mo6S8 chevrel phase cathode for rechargeable magnesium battery[J]. Journal of the Electrochemical Society, 2014, 161(4): A593-A598.
[89] LANCRY E, LEVI E, GOFER Y, et al. Leaching chemistry and the performance of the Mo6S8 cathodes in rechargeable Mg batteries[J]. Chemistry of Materials, 2004, 16(14): 2832-2838.
[90] LEVI E, GERSHINSKY G, AURBACH D, et al. New insight on the unusually high ionic mobility in chevrel phases[J]. Chemistry Materials, 2009, 21(7): 1390-1399.
[91] LEVI E, LANCRY E, MITELMAN A, et al. Phase diagram of Mg insertion into Chevrel phases, MgxMo6T8 (T=S, Se). 1. Crystal structure of the sulfides[J]. Chemistry of Materials, 2006, 18(23): 5492-5503.
[92] YOO H D, SHTERENBERG I, GOFER Y, et al. Mg rechargeable batteries: An on-going challenge[J]. Energy & Environmental Science, 2013, 6(8): 2265-2279
[93] WAN L F, PERDUE B R, APBLETT C A, et al. Mg desolvation and intercalation mechanism at the Mo6S8 chevrel phase surface[J]. Chemistry of Materials, 2015, 27(17): 5932-5940.
[94] LIANG Y, YOO H D, LI Y, et al. Interlayer-expanded molybdenum disulfide nanocomposites for electrochemical magnesium storage[J]. Nano Letters, 2015, 15(3): 2194-2202.
[95] YOO H D, LIANG Y, DONG H, et al. Fast kinetics of magnesium monochloride cations in interlayer-expanded titanium disulfide for magnesium rechargeable batteries[J]. Nature Communications, 2017, 8(1): 339.
[96] SHEN Y, WANG Y, MIAO Y, et al. High-energy interlayer- expanded copper sulfide cathode material in non-corrosive electrolyte for rechargeable magnesium batteries[J]. Advanced Materials, 2020, 32(4): 1905524.
[97] SUN X, BONNICK P, DUFFORT V, et al. A high capacity thiospinel cathode for Mg batteries[J]. Energy & Environmental Science, 2016, 9(7): 2273-2277.
[98] LIU M, JAIN A, RONG Z, et al. Evaluation of sulfur spinel compounds for multivalent battery cathode applications[J]. Energy & Environmental Science, 2016, 9(10): 3201-3209.
[99] DEY S, LEE J, BRITTO S, et al. Exploring cation-anion redox processes in one-dimensional linear chain vanadium tetrasulfide rechargeable magnesium ion cathodes[J]. Journal of the American Chemical Society, 2020, 142(46): 19588-19601.
[100] LI Z, VINAYAN B P, JANKOWSKI P, et al. Multi-electron reactions enabled by anion-based redox chemistry for high- energy multivalent rechargeable batteries[J]. Angewandte Chemie International Edition, 2020, 59(28): 11483-11490.
[101] MAO M, TONG Y, ZHANG Q, et al. Joint cationic and anionic redox chemistry for advanced Mg batteries[J]. Nano Letters, 2020, 20(9): 6852-6858.
[102] LI H, MA L, HAN C, et al. Advanced rechargeable zinc-based batteries: Recent progress and future perspectives[J]. Nano Energy, 2019, 62: 550-587.
[103] NOVK P, SHKLOVER V, NESPER R. Magnesium insertion in vanadium oxides: A structural study[J]. Zeitschrift für Physikalische Chemie, 1994, 185(1): 51-68.
[104] FU Q, SARAPULOVA A, TROUILLET V, et al. In operando synchrotron diffraction and in operando X-ray absorption spectroscopy investigations of orthorhombic V2O5 nanowires as cathode materials for Mg-ion batteries[J]. Journal of the American Chemical Society, 2019, 141(6): 2305-2315.
[105] PEREIRA-RAMOS J P, MESSINA R, PERICHON J. Electrochemical formation of a magnesium vanadium bronze MgxV2O5 in sulfone-based electrolytes at 150 ℃[J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1987, 218(1): 241-249.
[106] LEVI E, GOFER Y, AURBACH D. On the way to rechargeable Mg batteries: The challenge of new cathode materials[J]. Chemistry of Materials, 2010, 22(3): 860-868.
[107] NOVK P, IMHOF R, HAAS O. Magnesium insertion electrodes for rechargeable nonaqueous batteries—A competitive alternative to lithium?[J]. Electrochimica Acta, 1999, 45(1/2): 351-367.
[108] KRISTOFFERSEN H H, METIU H. Structure of V2O5·nH2O xerogels[J]. The Journal of Physical Chemistry C, 2016, 120(7): 3986-3992.
[109] SENGUTTUVAN P, HAN S D, KIM S, et al. A high power rechargeable nonaqueous multivalent Zn/V2O5 battery[J]. Advanced Energy Materials, 2016, 6(24): 1600826.
[110] LE D B, PASSERINI S, COUSTIER F, et al. Intercalation of polyvalent cations into V2O5 aerogels[J]. Chemistry of Materials, 1998, 10(3): 682-684.
[111] TEPAVCEVIC S, LIU Y, ZHOU D, et al. Nanostructured layered cathode for rechargeable Mg-ion batteries[J]. ACS Nano, 2015, 9(8): 8194-8205.
[112] SA N, KINNIBRUGH T L, WANG H, et al. Structural evolution of reversible Mg insertion into a bilayer structure of V2O5·nH2O xerogel material[J]. Chemistry of Materials, 2016, 28(9): 2962-2969.
[113] RASTGOO-DEYLAMI M, CHAE M S, HONG S T. H2V3O8 as a high energy cathode material for nonaqueous magnesium-ion batteries[J]. Chemistry of Materials, 2018, 30(21): 7464-7472.
[114] WANG R, CHUNG C C, LIU Y, et al. Electrochemical intercalation of Mg2+ into anhydrous and hydrated crystalline tungsten oxides[J]. Langmuir, 2017, 33(37): 9314-9323.
[115] WANG R, BOYD S, BONNESEN P V, et al. Effect of water in a non-aqueous electrolyte on electrochemical Mg2+ insertion into WO3[J]. Journal of Power Sources, 2020, 477: 229015.
[116] SAI GAUTAM G, CANEPA P, RICHARDS W D, et al. Role of structural H2O in intercalation electrodes: The case of Mg in nanocrystalline xerogel-V2O5[J]. Nano Letters, 2016, 16(4): 2426-2431.
[117] SA N, WANG H, PROFFIT D L, et al. Is alpha-V2O5 a cathode material for Mg insertion batteries?[J]. Journal of Power Sources, 2016, 323: 44-50.
[118] LOPEZ M, YOO H D, HU L, et al. Does water enhance Mg intercalation in oxides? The case of a tunnel framework[J]. ACS Energy Letters, 2020, 5(11): 3357-3361.
[119] ESPARCIA E A Jr, CHAE M S, OCON J D, et al. Ammonium vanadium bronze (NH4V4O10) as a high-capacity cathode material for nonaqueous magnesium-ion batteries[J]. Chemistry of Materials, 2018, 30(11): 3690-3696.
[120] GAUTAM G S, CANEPA P, MALIK R, et al. First- principles evaluation of multi-valent cation insertion into orthorhombic V2O5[J]. Chemical Communications, 2015, 51(71): 13619-13622.
[121] KITCHAEV D A, DACEK S T, SUN W, et al. Thermodynamics of phase selection in MnO2 framework structures through alkali intercalation and hydration[J]. Journal of the American Chemical Society, 2017, 139(7): 2672-2681.
[122] SPAHR M E, NOVAK P, HAAS O, et al. Electrochemical insertion of lithium, sodium, and magnesium in molybdenum(Ⅵ) oxide[J]. Journal of Power Sources, 1995, 54(2): 346-351.
[123] GERSHINSKY G, YOO H D, GOFER Y, et al. Electrochemical and spectroscopic analysis of Mg2+ intercalation into thin film electrodes of layered oxides: V2O5 and MoO3[J]. Langmuir, 2013, 29(34): 10964-10972.
[124] INCORVATI J T, WAN L F, KEY B, et al. Reversible magnesium intercalation into a layered oxyfluoride cathode[J]. Chemistry of Materials, 2016, 28(1): 17-20.
[125] KAVEEVIVITCHAI W, JACOBSON A J. High capacity rechargeable magnesium-ion batteries based on a microporous molybdenum-vanadium oxide cathode[J]. Chemistry of Materials, 2016, 28(13): 4593-4601.
[126] ZHANG M, MACRAE A C, LIU H, et al. Communication—Investigation of anatase-TiO2 as an efficient electrode material for magnesium-ion batteries[J]. Journal of the Electrochemical Society, 2016, 163(10): A2368-A2370.
[127] KOKETSU T, MA J, MORGAN B J, et al. Reversible magnesium and aluminium ions insertion in cation-deficient anatase TiO2[J]. Nature Materials, 2017, 16(11): 1142-1148.
[128] WANG Y, XUE X, LIU P, et al. Atomic substitution enabled synthesis of vacancy-rich two-dimensional black TiO2-x nanoflakes for high-performance rechargeable magnesium batteries[J]. ACS Nano, 2018, 12(12): 12492-12502.
[129] LUO L, ZHOU K, LIAN R, et al. Cation-deficient TiO2(B) nanowires with protons charge compensation for regulating reversible magnesium storage[J]. Nano Energy, 2020, 72: 104716.
[130] YANG J, LI J, GONG W, et al. Genuine divalent magnesium-ion storage and fast diffusion kinetics in metal oxides at room temperature[J]. Proceedings of the National Academy of Sciences, 2021, 118(38): e2111549118.
[131] BRUCE P G, KROK F, NOWINSKI J, et al. Chemical intercalation of magnesium into solid hosts[J]. Journal of Materials Chemistry, 1991, 1(4): 705-706.
[132] ZHANG R, YU X, NAM K-W, et al. α-MnO2 as a cathode material for rechargeable Mg batteries[J]. Electrochemistry Communications, 2012, 23: 110-113.
[133] ARTHUR T S, ZHANG R, LING C, et al. Understanding the electrochemical mechanism of K-αMnO2 for magnesium battery cathodes[J]. ACS Applied Materials & Interfaces, 2014, 6(10): 7004-7008.
[134] NAM K W, KIM S, LEE S, et al. The high performance of crystal water containing manganese birnessite cathodes for magnesium batteries[J]. Nano Letters, 2015, 15(6): 4071-4079.
[135] KIM S, NAM K W, LEE S, et al. Direct observation of an anomalous spinel-to-layered phase transition mediated by crystal water intercalation[J]. Angewandte Chemie International Edition, 2015, 54(50): 15094-15099.
[136] KIM C, PHILLIPS P J, KEY B, et al. Direct observation of reversible magnesium ion intercalation into a spinel oxide host[J]. Advanced Materials, 2015, 27(22): 3377-3384.
[137] BAYLISS R D, KEY B, SAI GAUTAM G, et al. Probing Mg migration in spinel oxides[J]. Chemistry of Materials, 2020, 32(2): 663-670.
[138] LIU M, RONG Z, MALIK R, et al. Spinel compounds as multivalent battery cathodes: A systematic evaluation based on ab initio calculations[J]. Energy & Environmental Science, 2015, 8(3): 964-974.
[139] SINHA N N, MUNICHANDRAIAH N. Electrochemical conversion of LiMn2O4 to MgMn2O4 in aqueous electrolytes[J]. Electrochemical and Solid-State Letters, 2008, 11: F23-F26.
[140] OKAMOTO S, ICHITSUBO T, KAWAGUCHI T, et al. Intercalation and push-out process with spinel-to-rocksalt transition on Mg insertion into spinel oxides in magnesium batteries[J]. Advanced Science, 2015, 2(8): 1500072.
[141] SAI GAUTAM G, CANEPA P, URBAN A, et al. Influence of inversion on Mg mobility and electrochemistry in spinels[J]. Chemistry of Materials, 2017, 29(18): 7918-7930.
[142] KWON B J, LAU K C, PARK H, et al. Probing electrochemical Mg-ion activity in MgCr2–xVxO4 spinel oxides[J]. Chemistry of Materials, 2019, 32(3): 1162-1171.
[143] CHEN T, SAI GAUTAM G, HUANG W, et al. First-principles study of the voltage profile and mobility of Mg intercalation in a chromium oxide spinel[J]. Chemistry of Materials, 2018, 30(1): 153-162.
[144] HU L H, JOHNSON I D, KIM S, et al. Tailoring the electrochemical activity of magnesium chromium oxide towards Mg batteries through control of size and crystal structure[J]. Nanoscale, 2019, 11(2): 639-646.
[145] KWON B J, YIN L, PARK H, et al. High voltage Mg-ion battery cathode via a solid solution Cr-Mn spinel oxide[J]. Chemistry of Materials, 2020, 32(15): 6577-6587.
[146] YIN L, KWON B J, CHOI Y, et al. Operando X-ray diffraction studies of the Mg-ion migration mechanisms in spinel cathodes for rechargeable Mg-ion batteries[J]. Journal of the American Chemical Society, 2021, 143(28): 10649-10658.
[147] HU L H, JOKISAARI J R, KWON B J, et al. High capacity for Mg2+ deintercalation in spinel vanadium oxide nanocrystals[J]. ACS Energy Letters, 2020, 5(8): 2721-2727.
[148] SA N, MUKHERJEE A, HAN B, et al. Direct observation of MgO formation at cathode electrolyte interface of a spinel MgCo2O4 cathode upon electrochemical Mg removal and insertion[J]. Journal of Power Sources, 2019, 424: 68-75.
[149] WANGOH L W, YANG Z Z, WANG L, et al. Mg2+ diffusion-induced structural and property evolution in epitaxial Fe3O4 thin films[J]. ACS Nano, 2020, 14(11): 14887-14894.
[150] SHIMOKAWA K, ATSUMI T, HARADA M, et al. Zinc-based spinel cathode materials for magnesium rechargeable batteries: Toward the reversible spinel-rocksalt transition[J]. Journal of Materials Chemistry A, 2019, 7(19): 12225-12235.
[151] SHIMOKAWA K, ATSUMI T, OKAMOTO N L, et al. Structure design of long-life spinel-oxide cathode materials for magnesium rechargeable batteries[J]. Advanced Materials, 2021, 33(7): 2007539.
[152] WUSTROW A, KEY B, PHILLIPS P J, et al. Synthesis and characterization of MgCr2S4 thiospinel as a potential magnesium cathode[J]. Inorganic Chemistry, 2018, 57(14): 8634-8638.
[153] MIURA A, ITO H, BARTEL C J, et al. Selective metathesis synthesis of MgCr2S4 by control of thermodynamic driving forces[J]. Materials Horizons, 2020, 7(5): 1310-1316.
[154] ZHANG Y, KONYA M, KUTSUMA A, et al. Magnesium storage performance and mechanism of 2D-ultrathin nanosheet-assembled spinel MgIn2S4 cathode for high-temperature Mg batteries[J]. Small, 2019, 15(36): 1902236.
[155] CANEPA P, BO S H, SAI GAUTAM G, et al. High magnesium mobility in ternary spinel chalcogenides[J]. Nature Communications, 2017, 8(1): 1759.
[156] 李 钊, 王 忠, 李 强, 等. 一种碳纳米管改性富锂锰基正极材料的策略[J]. 无机化学学报, 2019, 35(9): 1561-1569.
LI Zhao, WANG Zhong, LI Qiang, et al. A strategy for carbon nanotubes modified lithium-manganese-rich cathode material[J]. Chinese Journal of Inorganic Chemistry, 2019, 35(9): 1561-1569.
[157] LU S J, LIU Y, HE Z J, et al. Synthesis and properties of single-crystal Ni-rich cathode materials in Li-ion batteries[J]. Transactions of Nonferrous Metals Society of China, 2021, 31(4): 1074-1086.
[158] ISHIDA N, YAMAZAKI N, MANDAI T, et al. Crystal structures and cathode properties of chemically and electrochemically delithiated LixNi0.5Mn0.5O2 with applications to Mg rechargeable batteries[J]. Journal of the Electrochemical Society, 2020, 167(10): 100547.
[159] ISHIDA N, NISHIGAMI R, KITAMURA N, et al. Crystal structure analysis and electrochemical properties of chemically delithiated Li0.13Mn0.54Ni0.13Co0.13O2-δ as cathode material for rechargeable Mg batteries[J]. Chemistry Letters, 2017, 46(10): 1508-1511.
[160] ISHIDA N, NISHIGAMI R, MATSUI M, et al. Revisiting delithiated Li1.2Mn0.54Ni0.13Co0.13O2: Structural analysis and cathode properties in magnesium rechargeable battery applications[J]. Electrochemistry, 2021, 89(4): 329-333. doi:10.5796/ electrochemistry.21-00038.
[161] 曾 涛, 安长胜, 易 旭, 等. 石墨烯引导LiFePO4纳米片的一步溶剂热反应制备及其电化学性能[J]. 中国有色金属学报, 2019, 29(2): 319-325.
ZENG Tao, AN Chang-sheng, YI Xu, et al. Preparation and enhanced electrochemical performance of LiFePO4 nanoflakes directed by graphene through one-pot solvothermal reaction[J]. The Chinese Journal of Nonferrous Metals, 2019, 29(2): 319-325.
[162] LING C, BANERJEE D, SONG W, et al. First-principles study of the magnesiation of olivines: Redox reaction mechanism, electrochemical and thermodynamic properties[J]. Journal of Materials Chemistry, 2012, 22(27): 13517-13523.
[163] LE POUL N, BAUDRIN E, MORCRETTE M, et al. Development of potentiometric ion sensors based on insertion materials as sensitive element[J]. Solid State Ionics, 2003, 159(1/2): 149-158.
[164] ZHANG R, LING C. Unveil the chemistry of olivine FePO4 as magnesium battery cathode[J]. ACS Applied Materials & Interfaces, 2016, 8(28): 18018-18026.
[165] ORIKASA Y, KISU K, IWAMA E, et al. Noncrystalline nanocomposites as a remedy for the low diffusivity of multivalent ions in battery cathodes[J]. Chemistry of Materials, 2020, 32(3): 1011-1021.
[166] ORIKASA Y, MASESE T, KOYAMA Y, et al. High energy density rechargeable magnesium battery using earth- abundant and non-toxic elements[J]. Scientific Reports, 2014, 4(1): 5622.
[167] HEATH J, CHEN H, ISLAM M S. MgFeSiO4 as a potential cathode material for magnesium batteries: ion diffusion rates and voltage trends[J]. Journal of Materials Chemistry A, 2017, 5(25): 13161-13167.
[168] NULI Y, YANG J, WANG J, et al. Electrochemical intercalation of Mg2+ in magnesium manganese silicate and its application as high-energy rechargeable magnesium battery cathode[J]. The Journal of Physical Chemistry C, 2009, 113(28): 12594-12597.
[169] FENG Z, YANG J, NULI Y, et al. Preparation and electrochemical study of a new magnesium intercalation material Mg1.03Mn0.97SiO4[J]. Electrochemistry Communications, 2008, 10(9): 1291-1294.
[170] TORRES A, ARROYO-DE DOMPABLO M E. Comparative investigation of MgMnSiO4 and olivine-type MgMnSiS4 as cathode materials for Mg batteries[J]. The Journal of Physical Chemistry C, 2018, 122(17): 9356-9362.
[171] MORI T, MASESE T, ORIKASA Y, et al. Anti-site mixing governs the electrochemical performances of olivine-type MgMnSiO4 cathodes for rechargeable magnesium batteries[J]. Physical Chemistry Chemical Physics, 2016, 18(19): 13524-13529.
[172] ZHENG Y, NULI Y, CHEN Q, et al. Magnesium cobalt silicate materials for reversible magnesium ion storage[J]. Electrochimica Acta, 2012, 66: 75-81.
[173] SALAMA M, ROSY, ATTIAS R, et al. Metal-sulfur batteries: Overview and research methods[J]. ACS Energy Letters, 2019, 4(2): 436-446.
[174] 李英楠, 闫晓燕, 刘宝胜, 等. 金属硫化物在锂硫电池正极中的应用进展[J]. 中国有色金属学报, 2021, 31(11): 3272-3288.
LI Ying-nan, YAN Xiao-yan, LIU Bao-sheng, et al. Application progress of metal sulfides materials forlithium- sulfur batteries cathode[J]. The Chinese Journal of Nonferrous Metals, 2021, 31(11): 3272-3288.
[175] GAO T, JI X, HOU S, et al. Thermodynamics and kinetics of sulfur cathode during discharge in MgTFSI2-DME electrolyte[J]. Advanced Materials, 2018, 30(3): 1704313.
[176] ZHANG T, TAO Z, CHEN J. Magnesium-air batteries: From principle to application[J]. Materials Horizons, 2014, 1(2): 196-206.
[177] SHIGA T, HASE Y, KATO Y, et al. A rechargeable non-aqueous Mg-O2 battery[J]. Chemical Communications, 2013, 49(80): 9152-9154.
[178] TIAN H, GAO T, LI X, et al. High power rechargeable magnesium/iodine battery chemistry[J]. Nature Communications, 2017, 8(1): 14083.
[179] BITENC J, PIRNAT K, BANCIC T, et al. Anthraquinone- based polymer as cathode in rechargeable magnesium batteries[J]. Chem Sus Chem, 2015, 8(24): 4128-4132.
[180] LIANG Y, JING Y, GHEYTANI S, et al. Universal quinone electrodes for long cycle life aqueous rechargeable batteries[J]. Nature Materials, 2017, 16(8): 841-848.
[181] DONG H, LIANG Y, TUTUSAUS O, et al. Directing Mg-storage chemistry in organic polymers toward high- energy Mg batteries[J]. Joule, 2019, 3(3): 782-793.
[182] SUN X, ZHANG X, LIU W, et al. Electrochemical performances and capacity fading behaviors of activated carbon/hard carbon lithium ion capacitor[J]. Electrochimica Acta, 2017, 235: 158-166.
[183] 李 钊, 孙现众, 李 晨, 等. 介孔石墨烯/炭黑复合导电剂在锂离子电容器负极中的应用[J]. 储能科学与技术, 2017, 6(6): 1264-1272.
LI Zhao, SUN Xian-zhong, LI Chen, et al. Application of mesoporous graphene/carbon black composite conductive additive in lithium-ion capacitor anode[J]. Energy Storage Science and Technology, 2017, 6(6): 1264-1272.
[184] 李 钊, 孙现众, 刘文杰, 等. 预嵌锂硬碳和软碳用于锂离子电容器负极的比较研究(英文)[J]. 电化学, 2019, 25(1): 122-136.
LI Zhao, SUN Xian-zhong, LIU Wen-Jie, et al. A comparative study of pre-lithiated hard carbon and soft carbon as anodes for lithium-ion capacitors[J]. Journal of Electrochemistry, 2019, 25(1): 122-136.
[185] YOO H D, HAN S D, BAYLISS R D, et al. “Rocking-Chair”-type metal hybrid supercapacitors[J]. ACS Applied Materials & Interfaces, 2016, 8(45): 30853-30862.
[186] ATTIAS R, SHARON D, GOFFER Y, et al. Critical review on the unique interactions and electroanalytical challenges related to cathodes-solutions interfaces in non-aqueous Mg battery prototypes[J]. Chem Electro Chem, 2021, 8(17): 3229-3238.
[187] ZHANG J, GUAN X, LV R, et al. Rechargeable Mg metal batteries enabled by a protection layer formed in vivo[J]. Energy Storage Materials, 2020, 26: 408-413.
[188] WAN B, DOU H, ZHAO X, et al. Three-dimensional magnesiophilic scaffolds for reduced passivation toward high-rate Mg metal anodes in a noncorrosive electrolyte[J]. ACS Applied Materials & Interfaces, 2020, 12(25): 28298-28305.
[189] MANDAI T, SOMEKAWA H. Metallurgical approach to enhance the electrochemical activity of magnesium anodes for magnesium rechargeable batteries[J]. Chemical Communications, 2020, 56(81): 12122-12125.
[190] ARTHUR T S, SINGH N, MATSUI M. Electrodeposited Bi, Sb and Bi1-xSbx alloys as anodes for Mg-ion batteries[J]. Electrochemical Communications, 2012, 16(1): 103-106.
[191] SINGH N, ARTHUR T S, LING C, et al. A high energy- density tin anode for rechargeable magnesium-ion batteries[J]. Chemical Communications, 2013, 49(2): 149-151.
[192] MURGIA F, WELDEKIDAN E T, STIEVANO L, et al. First investigation of indium-based electrode in Mg battery[J]. Electrochemical Communications, 2015, 60: 56-59.
[193] MURGIA F, MONCONDUIT L, STIEVANO L, et al. Electrochemical magnesiation of the intermetallic InBi through conversion-alloying mechanism[J]. Electrochimica Acta, 2016, 209: 730-736.
[194] NGUYEN D T, SONG S W. Magnesium stannide as a high-capacity anode for magnesium-ion batteries[J]. Journal of Power Sources, 2017, 368: 11-17.
[195] TAN Y H, YAO W T, ZHANG T, et al. High voltage magnesium-ion battery enabled by nanocluster Mg3Bi2 alloy anode in noncorrosive electrolyte[J]. ACS Nano, 2018, 12(6): 5856-5865.
[196] MENG Z, FOIX D, BRUN N, et al. Alloys to replace Mg anodes in efficient and practical Mg-ion/sulfur batteries[J]. ACS Energy Letters, 2019, 4(9): 2040-2044.
[197] SHAO Y, GU M, LI X, et al. Highly reversible Mg insertion in nanostructured Bi for Mg ion batteries[J]. Nano Letters, 2014, 14(1): 255-260.
[198] ZHANG D, FU J, WANG Z, et al. Perspective—Reversible magnesium storage in silicon: An ongoing challenge[J]. Journal of The Electrochemical Society, 2020, 167(5): 050514.
[199] GOD C, BITSCHNAU B, KAPPER K, et al. Intercalation behaviour of magnesium into natural graphite using organic electrolyte systems[J]. RSC Advances, 2017, 7(23): 14168-14175.
[200] KIM D M, JUNG S C, HA S, et al. Cointercalation of Mg2+ ions into graphite for magnesium-ion batteries[J]. Chemistry of Materials, 2018, 30(10): 3199-3203.
[201] KAWAGUCHI M, KURASAKI A. Intercalation of magnesium into a graphite-like layered material of composition BC2N[J]. Chemical Communications, 2012, 48(55): 6897-6899.
[202] ER D, DETSI E, KUMAR H, et al. Defective graphene and graphene allotropes as high-capacity anode materials for Mg ion batteries[J]. ACS Energy Letters, 2016, 1(3): 638-645.
[203] WU N, YIN Y X, GUO Y G. Size-dependent electrochemical magnesium storage performance of spinel lithium titanate[J]. Chemistry—An Asian Journal, 2014, 9(8): 2099-2102.
[204] WU N, YANG Z Z, YAO H R, et al. Improving the electrochemical performance of the Li4Ti5O12 electrode in a rechargeable magnesium battery by lithium-magnesium co-intercalation[J]. Angewandte Chemie International Edition, 2015, 54(19): 5757-5761.
[205] SON S B, GAO T, HARVEY S P, et al. An artificial interphase enables reversible magnesium chemistry in carbonate electrolytes[J]. Nature Chemistry, 2018, 10(5): 532-539.
LI Zhao1, 2, 3, YAO Yin-yin1, 2, 3, LI Bo-fei4, WANG Li-chen1, 2, 3, XU Hao1, 2, 3, CHONG Li-na1, 2, 3, ZOU Jian-xin1, 2, 3
(1. School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China;
2. National Engineering Research Center of Light Alloy Net Forming, Shanghai Jiao Tong University, Shanghai, 200240, China;
3. Center of Hydrogen Science, Shanghai Jiao Tong University, Shanghai 200240, China;
4. School of Materials Engineering, Shanghai University of Engineering Science, Shanghai 201620, China)
Abstract: Rechargeable magnesium batteries (RMBs) are expected to be the preferred electrochemical devices for large-scale energy storage technology under the vision of achieving carbon peak and neutrality goals. RMBs have not been commercialized due to some key scientific questions and technical bottlenecks remain unresolved. In this review, we introduced the advantages of safety and energy density of RMBs, and summarized the research history and advances of electrolytes, cathode and anode materials in RMBs. The significant role of Grignard-based, sulfonic acid and boron-based electrolytes in improving reversible deposition/dissolution as well as voltage window of magnesium are discussed carefully. Meanwhile, the mechanism of magnesium ion storage of Chevrel phase Mo6S8, sulfides and oxides are analyzed in detail. Then the intercalation cathode materials with high voltage (such as spinel, layered and polyanion structure materials), conversion cathode materials with high capacity(such as sulfur, oxygen and organic molecules) and activated carbon cathode with high power are introduced and discussed. In addition, metals such as magnesium, magnesium alloys, bismuth and tin as well as graphite et al. as anode are discussed from electrolyte/electrode interface reaction mechanism. Finally, the materials and devices design and application scenarios of practical RMBs is summarized and prospected.
Key words: rechargeable magnesium batteries; electrolytes; cathode materials; anode materials; development and challenge
Foundation item: Project(51771112) supported by the National Natural Science Foundation of China
Received date: 2021-09-30; Accepted date: 2021-11-08
Corresponding author: ZOU Jian-xin; Tel: +86-15921793455; E-mail: zoujx@sjtu.edu.cn
(编辑 何学锋)
基金项目:国家自然科学基金资助项目(51771112)
收稿日期:2021-09-30;修订日期:2021-11-08
通信作者:邹建新,教授,博士;电话:15921793455;E-mail:zoujx@sjtu.edu.cn
摘 要:可充镁电池因其高体积比容量、高安全性及原料镁储量丰富等优势,有望成为“双碳”目标下规模化储能技术的优选电化学器件。然而目前可充镁电池还未能实现商业化,这与其存在的一些关键科学问题尚未明晰以及技术瓶颈还未被突破等因素有关。因此,本文从可充镁电池的安全性和能量密度出发,在梳理了可充镁电池发展历史的基础上,总结了可充镁电池器件中电解液、正极和负极材料的研究进展。文中主要介绍了格氏基、磺酸基和硼基电解液对改善镁可逆沉积/溶解和提高电压窗口的重要作用,并对Chevrel相Mo6S8、硫化物和氧化物作为正极材料的储镁机制进行了详细分析。然后对高电压的插层正极材料(尖晶石、层状和聚阴离子化合物)、高容量的转化正极材料(硫、氧和有机分子)和高功率的活性炭正极进行了着重介绍。此外,从电解液/电极界面反应机制着手,对镁、镁合金、铋和锡等金属以及石墨等负极材料进行了梳理分析。最后,本文从材料设计、器件匹配和应用场景角度,对可充镁电池未来商业化的挑战进行了总结和展望。