Trans. Nonferrous Met. Soc. China 31(2021) 3342-3355
First-principles investigation on stability and electronic structure of Sc-doped θ′/Al interface in Al-Cu alloys
Dong-lan ZHANG1, Jiong WANG1, Yi KONG1, You ZOU2, Yong DU1
1. Powder Metallurgy Research Institute, Central South University, Changsha 410083, China;
2. Information and Network Center, Central South University, Changsha 410083, China
Received 14September 2021; accepted 3November 2021
Abstract:
he properties of Sc-doped θ′ (Al2Cu)/Al interface in Al-Cu alloys were investigated by first-principles calculations. Sc-doped semi-coherent and coherent θ′ (Al2Cu)/Al interfaces (Sc doped in Al slab (S1 site), Sc doped in θ′ slab (S2 site)) were modeled based on calculated results and reported experiments. Through the analysis of interfacial bonding strength, it is revealed that the doping of Sc at S1 site can significantly decrease the interface energy and increase the work of adhesion. In particular, the doped coherent interface with Sc at S1 site which is occupied by interstitial Cu atoms has very good bonding strength. The electronic structure shows the strong Al—Cu bonds at the interfaces with Sc at S1 site, and the Al—Al bonds at the interfaces with Sc at S2 site are formed. The formation of strong Al—Cu and Al—Al bonds plays an important role in the enhancement of doped interface strength.
Key words:
Al-Cu alloys;Sc-doped θ′/Al interface; interfacial bonding strength; electronic structure;
1 Introduction
Al-Cu alloys are widely used in automotive industry and aircraft applications as the popular lightweight engineering material [1-3]. Compared with steel, the lower strength of Al-Cu alloys has impeded their development in certain engineering applications. Precipitation strengtheningis the main means to make the alloys obtain extremely high strength and good comprehensive mechanical properties [4-6]. During the aging treatment, a series of nano-scale intermediate precipitates were formed by the decomposition of rapidly cooled supersaturated solid solutions. The main precipitation sequence widely accepted for Al-Cu alloys is: GP (Guinier–Preston) I zones → θ′′(GP II zones) → θ′ → θ[7-10]. The θ′(Al2Cu) phase is thermodynamically unstable and will transform to stable phase θ at 523-643 K, or melted at 703 K [11-14]. The presence of θ phase which is completely incoherent with the Al matrix will reduce the mechanical properties of the Al-Cu alloys. On the contrary, the metastable phase θ′ is coherent or semi-coherent with matrix, one of the main strengthening precipitate phases in Al-Cu alloys [6,15,16], with the high aspect ratio plate-like morphology which is closely related to interface structure [17,18]. It is well known that the interface between precipitation phases and Al matrix plays a vital role in Al-Cu alloys [19]. It can not only hinder the movement of dislocations and increase the strength of the material, but also act as potential nucleation sites for dislocations and increase plasticity [3]. For this reason, improving the stability of θ′ phase at high temperature and the stability of interface between θ′ and Al matrix is of great significance to obtain Al-Cu alloys with good comprehensive properties.
A great deal of work by experiments has been done to explore the effect of trace elements onθ′ phase in Al-Cu alloys. It is proved that trace elements of Au, Ag, Si and Sn are active in promoting the heterogeneous nucleation of metastable phase θ′[16,20-22], Pr, Zr and Cr can significantly inhibit the recrystallization and a large number of fine sub-grain boundaries are obtained in the Al-Cu alloys [23,24], and Sc, Mn and Zr can improve the thermal stability of the metastable phase θ′ through segregating at the interface between θ′ and Al matrix [25-27]. The addition of trace elements also has an important impact on the interface. YANG et al [28] and GAO et al [29] studied the Al-Cu alloys with trace Sc addition and found that Sc segregation at the interface could reduce the interface energy and make the alloy have better mechanical properties. However, since the precipitated phases in Al-Cu alloys are of nanometer level, it is difficult to experimentally investigate the interfacial properties and the mechanism of the effect of trace elements on the interface between precipitated phases and Al matrix. First-principles calculations based on atomic scale are widely used in the study of materials [30-37]. ZHOU et al [3] simulated the interface of θ/Al by molecular dynamics and proved that interfaces with different terminals have a strong influence on the nucleation site of dislocations. KIM et al [38] investigated the interface stability of θ′/Al by density functional theory calculations and found that the coherent interface without occupancy of interstitial Cu atoms was equilibrium interface, which is a good explanation of the phenomenon observed in experiment. Therefore, it is critical to further explore the addition of trace elements on interfacial properties of Al-Cu alloys from atomic levels.
In this work, Sc-doped θ′(010)/Al(010) and θ′(001)/Al(001) (semi-coherent and coherent) interfaces with different terminations and configurations were modeled, the energy and bonding strength of doped interfaces were investigated by first-principles calculations. Through the calculation and investigation of pure interfaces and Sc-doped interfaces properties, our results will provide a more microscopic theoretical explanation for the experimental analysis at the atomic level and from an electronic perspective andprovide theoretical guidance for the study of the influence of minor Sc addition on the stability of θ′/Al interface and the design of trace element doped Al-Cu alloys.
2 Methodology
2.1 First-principles calculations
All calculations were performed with the VASP (Vienna Ab initio Simulation Package) [39] and ALKEMIE platform [40]. The PAW (projector augmented wave) potentials and the PBE (Perdew-Burke-Ernzerhof) function of the GGA (generalized gradient approximation) were used to treat the exchange-correlation effects [41].The BFGS (Broyden-Fletcher-Goldfarb-Shannon) algorithm [42-44] was used to relax all the bulk, surface and interface structures to obtain a stable system with the minimum total energy. The Brillouin zone was sampled with Monkhorst-Pack scheme [45] using the k-points grid of 18 × 18 × 18 for bulk, 9 × 9 × 1, and 5 × 15 × 1 for θ′(001)/Al(001) and θ′(010)/Al(010) interfaces calculations, respectively. The cut-off energy was set to be 400 eV for plane-wave expansions, and the thickness of vacuum layer was set to be 12.0 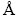 for all the interface models based on convergence test. The total energy was converged to 1.0×10-5 eV/atom during the optimization.
for all the interface models based on convergence test. The total energy was converged to 1.0×10-5 eV/atom during the optimization.
2.2 Surface properties
In order to simulate the surface of bulk, the slab model is used and should be guaranteed to exhibit bulk-like interiors. But the more the number of slab layers was used, the longer the calculation time was needed. Therefore, the surface convergence tests of slabs Al(001), Al(010), θ′(001) and θ′(010) were carried out at first. The surface energy (σ) is conventionally defined as the reversible energy needed to create a specific surface from a bulk unit cell, which can be used to describe the stability of the surfaces. The calculation formulas of σAl and σθ′areexpressed as Eq. (1) and Eq. (2) [46,47], respectively:
 (1)
(1)
where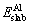 is the total energy of the Al slab with vacuum space, Ebulk is the total energy of the Al bulk, nslab and nbulk are the numbers of Al atom in the surface slab and the Al bulk material,respectively, and Asurfis the area of the surface.
is the total energy of the Al slab with vacuum space, Ebulk is the total energy of the Al bulk, nslab and nbulk are the numbers of Al atom in the surface slab and the Al bulk material,respectively, and Asurfis the area of the surface.
 (2)
(2)
where 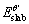 is the total energy of the θ′ surface slab with vacuum space, ECu is the energy of per Cu atom in the Cu bulk material,
is the total energy of the θ′ surface slab with vacuum space, ECu is the energy of per Cu atom in the Cu bulk material, 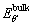 is the total energy of the θ′ bulk material, nCu andnAl are the numbers of Cu and Al atoms in the θ′ surface slab, respectively, and nθ′ is the number of θ′ formula in the θ′ bulk material. Equations (1) and (2) are only applicable to the stoichiometric surfaces.
is the total energy of the θ′ bulk material, nCu andnAl are the numbers of Cu and Al atoms in the θ′ surface slab, respectively, and nθ′ is the number of θ′ formula in the θ′ bulk material. Equations (1) and (2) are only applicable to the stoichiometric surfaces.
The curves of surface energy varying with the number of layers are shown in Fig. 1. It can be found that theσvalues of slab Al(001) and Al(010) converge well to 0.96 and 0.93 J/m2with six-layer slabs, which is in good accordance with the experimental value of 0.98 J/m2 [48]. The σvalueof slab θ′(010) tends to converge around 1.25 J/m2 with six-layer slabs. There are two different terminations for θ′(001) slabs, Al termination (θ′(001)-Al) and Cu termination (θ′(001)-Cu). The σ values of slab θ′(001)-Al and θ′(001)-Cu tend to converge around 1.50 J/m2 with six layer slabs, which is in good agreement with the calculation results by SUN et al [49] as shown in Fig. 1. It can be seen that the surface energy values of θ′ will still fluctuate slightly with more than six layer slabs.
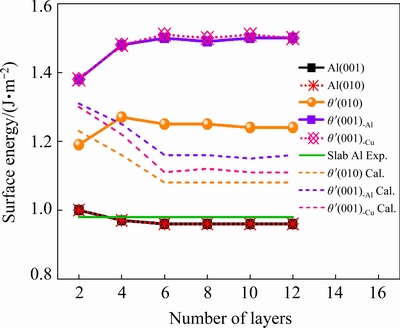
Fig. 1 Surface energy of Al and θ′(Al2Cu) slabs (The experimental value (solid line) is from Ref. [48] and the calculation values (dotted lines) are from Ref. [49])
In order to make the result more convincing, the change of interlayer spacing (△dij) on slabs of θ′(010), θ′(001)-Al and θ′(001)-Cu was taken into account. The △dij is defined as follows:
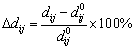 (3)
(3)
wheredij is the spacing between the adjacent layers i and j after relaxation, the 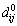 is the spacing before relaxation between layers i and j in an ideal crystal. As can be seen from Table 1, the change of interlayer spacing is obvious among the first three layers. Combined with Fig. 1, it can be fully confirmed that six layer slabs of θ′(010), θ′(001)-Al and θ′(001)-Cu are enough to exhibit bulk-like interior.
is the spacing before relaxation between layers i and j in an ideal crystal. As can be seen from Table 1, the change of interlayer spacing is obvious among the first three layers. Combined with Fig. 1, it can be fully confirmed that six layer slabs of θ′(010), θ′(001)-Al and θ′(001)-Cu are enough to exhibit bulk-like interior.
2.3 Properties of pure and doped interface
2.3.1 Interface models
Due to the difference in lattice parameters between the two different slabs, it is often necessary to consider the interface mismatch when an interface is constructed. It is generally believed that when the mismatch is less than 5%, the interface is more reliable [50]. The semi-coherent θ′(010)/Al(010) interface has a large interface mismatch of 18.3%, which can be reduced to 2.85% by expanding the slab along the a-direction with a≈3aAl≈2aθ′. It is worth noting that each layer of slab θ′(010) can be terminated by Al and Cu atoms, and the slab has symmetrical structure with seven layers, which can eliminate the influence of surface dipoles. On the contrary, the coherent θ′(001)/Al(001) interface with the lattice parameters a≈aAl≈aθ′ possesses a very small mismatch of 1.13%.Each layer of slab θ′(001) can be terminated by either Al or Cu atoms, so both terminals are considered. Taking into account the influence of interface spacing on the interface structure stability, the interface spacing tests of coherent and semi-coherent interfaces were performed. Finally, the equilibrium interface structures of two semi-coherent θ′(010)/Al(010) interfaces and four coherent θ′(001)/Al(001) interfaces with different terminations and configurations were modeled as shown in Figs. 2(a-f). It is interesting to note that both BOURGEOIS et al [15] and GAO et al [19] found that the interstitial sites within θ′ of the coherent θ′/Al interfaces were occupied by additional Cu atoms (Cu-rich) via high-resolution electron microscopy, the schematic diagram of the interface atomic structure is shown in Fig. 2(i). Therefore, the Cu-rich coherent θ′(001)/Al(001) interfaces were also taken into account, as shown in Figs. 2(g) and (h).
Table 1 Calculated change of interlayer spacing (△dij) on θ′(010) and θ′(001) surfaces with different terminations after relaxation
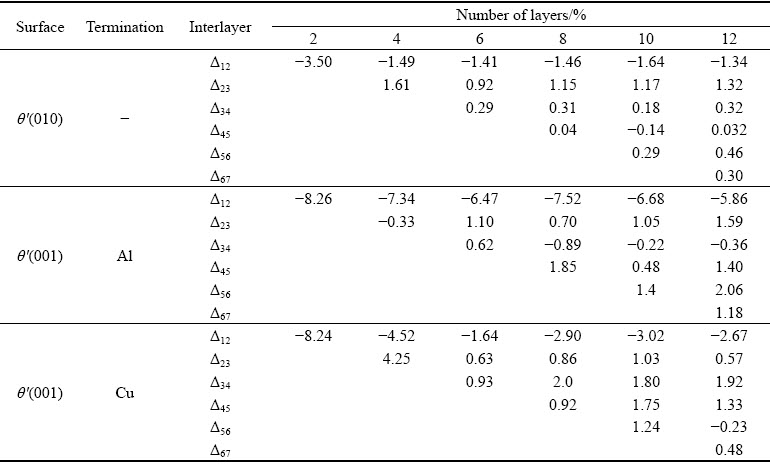

Fig. 2 Atomic structures of θ′/Al interfaces
2.3.2 Work of adhesion
The ideal work of adhesion (Wad) is the energy per unit area required to separate the interface into two free surfaces. It is related to the bonding strength of the interface and can be used to predict the mechanical properties of materials. The Wad is defined as follows [51,52]:
 (4)
(4)
where is the total energy of slab Al,
is the total energy of slab Al,  is the total energy of slab θ′,
is the total energy of slab θ′,  is the total energy of θ′/Al interface, and Aint is the total area of the interface.
is the total energy of θ′/Al interface, and Aint is the total area of the interface.
2.3.3 Interface energy of pure interface
Interface energy (γint) is the energy required to create an interface from bulk materials, and it is generally believed that the lower the interface energy, the more stable the interface. In order to build more stable doped interfaces later, it is necessary to consider the stability of the pure interfaces. The γint is defined as follows [53,54]:
 (5)
(5)
whereEint is the total energy of the fully relaxed interface, 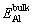 and are the total energies of single Al atom and θ′ molecular formula in the bulk material, respectively, nAland nθ′are the numbers of Al atom and θ′ molecular formula in the interface, respectively, and σAl and σθ′are the surface energies of Al and θ′ slabs, respectively.
and are the total energies of single Al atom and θ′ molecular formula in the bulk material, respectively, nAland nθ′are the numbers of Al atom and θ′ molecular formula in the interface, respectively, and σAl and σθ′are the surface energies of Al and θ′ slabs, respectively.
The results of all the considered interface configurations are listed in Table 2, and the γintvalues of the 1×1×1 unit cell and the 2×2×2 supercell of coherent θ′(001)/Al(001) interface are both calculated. For the coherent θ′(001)/Al(001) interface, the Cu-terminated interface with bridge site has the smallest interface energy, which is in good agreement with the experimental value [55], indicating that the coherent interface with Cu-terminated interface and bridge site stacking is the most stable. For the two semi-coherent θ′(010)/Al(010) interfaces with top and hollow site stacking respectively, the calculated results of γintare closer to the experimental data [55]than other theoretical values [5,38], indicating the reasonability of these two semi-coherent interfaces.
According to the analysis of the above results and the reported experimental phenomena [15,56], the four interfaces of θ′(001)-Cu/Al(001) with bridge site stacking, Cu-rich θ′(001)-Cu/Al(001) with bridge site stacking and θ′(010)/Al(010) with top and hollow site stacking were selected to build the Sc-doped interfaces. Considering two different doping situations of Sc atoms replacing Al atoms in slab Al (S1 site) or replacing Al atoms in slab θ′(S2 site), a total of eight Sc-doped interfaces were built, as shown in Fig. 3.
2.3.4 Interface energy of doped interface
Table 2 Interface energy (γint) of θ′(001)/Al(001) and θ′(010)/Al(010) interfaces with different terminations and configurations (mJ/m2)

Fig. 3 Atomic structures of doped θ′/Al interfaces
In order to further explore the effect of Sc doping on the interface thermodynamic stability, the γint of doped interfaces was also calculated. For non-stoichiometric compounds, the γint is related to the chemical potential of each phase [57], so Eq. (5) is no longer applicable. It can be defined by the following form [53,58]:
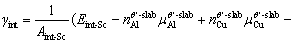
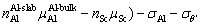 (6)
(6)
where 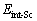 is the total energy of Sc-doped interface,
is the total energy of Sc-doped interface,  and
and 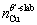 are the numbers of corresponding atoms in the θ′ slab of interface supercell,
are the numbers of corresponding atoms in the θ′ slab of interface supercell,  is the number of Al atoms in the Al slab of interface supercell, μAl, μCu andμSc are the chemical potentials of Al, Cu and Sc atoms, respectively, nSc is the number of Sc atoms in the interface supercell, Aint-Sc is the area of the Sc-doped interface,σAl and σθ′are the surface energies of Al and θ′ slabs with or without Sc atoms, respectively. Considering the fact that the chemical potential of the bulk θ′ constrains the values of
is the number of Al atoms in the Al slab of interface supercell, μAl, μCu andμSc are the chemical potentials of Al, Cu and Sc atoms, respectively, nSc is the number of Sc atoms in the interface supercell, Aint-Sc is the area of the Sc-doped interface,σAl and σθ′are the surface energies of Al and θ′ slabs with or without Sc atoms, respectively. Considering the fact that the chemical potential of the bulk θ′ constrains the values of 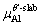 and
and 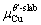 by Eq. (7):
by Eq. (7):
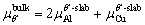 (7)
(7)
andsubstituting Eq. (7) into Eq. (6), the γint of the doped interfaces can be defined as follows:
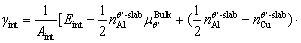
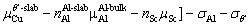 (8)
(8)
It can be seen from Eq. (8) that, the γint of doped interfaces depends on the chemical potential of Cu element;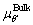 ,
, and
and  are thetotalenergies of θ′ formula, Al and Sc atoms in the bulk phases, respectively. For μCu, △μCu=μCu-
are thetotalenergies of θ′ formula, Al and Sc atoms in the bulk phases, respectively. For μCu, △μCu=μCu-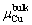 , and the value is constrained by Eq. (9):
, and the value is constrained by Eq. (9):
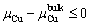 (9)
(9)
Considering the fact that the chemical potential of the bulk θ′ is also related to the heat of formation, the relationship is shown in Eq. (10):
 (10)
(10)
where△Hf is the heat of formation of bulk θ′, which is defined as follows [59,60]:
 (11)
(11)
where EAl and ECuare the energies of per Al and Cu atom, respectively, m and nare the numbers of corresponding Al and Cu atoms in the bulk θ′. The heat of formation of bulk θ′ was calculated to be -4.087 eV. Combining with Eqs. (7), (9) and (10), and supposing that the condition of poor Cu is 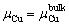 -4.087 (eV), the range for △μCu is given as
-4.087 (eV), the range for △μCu is given as
 (12)
(12)
It should be noted that when the Sc atoms are doped at S1 or S2 site, the surfaces of Al slab and θ′ slab are non-stoichiometric, Eq. (1) and Eq. (2) are no longer applicable for the calculation of σAl and σθ′. The surface energy σof Al and θ′ slabs with Sc atoms can be calculated according to Eqs. (13) and (14) for the S1 and S2 sites, respectively [46,53,61,62]:
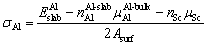 (13)
(13)
 (14)
(14)
wherenSc is the number of Sc atoms in Al or θ′ slab.
2.3.5 Segregation energy
The segregation energy (Eseg) is the energy required for the dopant to segregate at the interface, which is related to the interfacial strength. The more negative the value ofEseg, the stronger the ability of dopant atoms to segregate at the interface, and the more conducive to the bonding of the interface. The Eseg is defined as follows [53]:
 (15)
(15)
where the  and
and  are total energies of Sc-doped interface and pure interface, respectively,
are total energies of Sc-doped interface and pure interface, respectively, and are energies of a Sc atom and an Al atom in the bulk, respectively.
and are energies of a Sc atom and an Al atom in the bulk, respectively.
3 Results and discussion
3.1 Bulk calculations
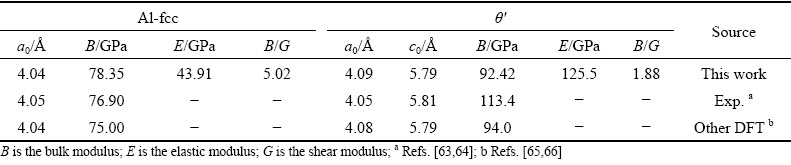
The crystal structure of Al bulk is cubic with the space group of 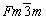 and θ′ bulkis tetragonal with the space group of I4/mmm, as shown in Fig. 4. The optimized lattice constants (a0, c0) and elastic moduli are listed in Table 3. It can be seen that our calculated results are in good agreement with the experimental values [63,64] and other theoretical calculations [65,66], indicating the reliability of the calculated parameters we adopted.
and θ′ bulkis tetragonal with the space group of I4/mmm, as shown in Fig. 4. The optimized lattice constants (a0, c0) and elastic moduli are listed in Table 3. It can be seen that our calculated results are in good agreement with the experimental values [63,64] and other theoretical calculations [65,66], indicating the reliability of the calculated parameters we adopted.
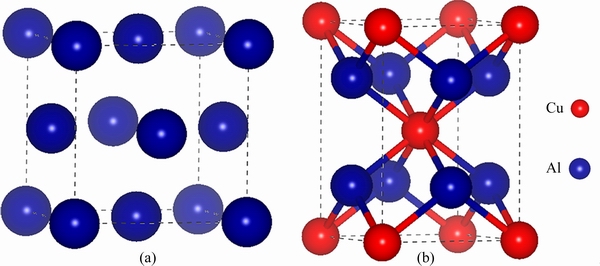
Fig. 4 Crystal structures of Al bulk (a) and θ′ (Al2Cu) bulk(b)
Table 3 Lattice constants a0, c0 and elastic moduli of Al bulk and θ′ (Al2Cu) bulk
3.2 Work of adhesion
The Wadvalues of the pure and doped interfaces are listed in Table 4. Compared with the pure interfaces, the Wad values of all the doped interfaces are increased except the θ′(010)/Al(010) interfaces with top site stacking and Sc at S2 site, and the Wad values of all the doped interfaces with Sc at S1 site are larger than those of all the doped interfaces with Sc at S2 site, indicating that Sc doping can be effective to improve the bonding strength of the θ′/Al interface, and the Sc atoms have a great tendency to be doped in the Al slab. It is worth noting that the Wad value of the Cu-rich θ′(001)-Cu/Al(001) interface with bridge site stacking and Sc at S1 site is much larger than that of all other interfaces, indicating that Sc doping has the greatest influence on the bonding strength of this interface, making it exhibit the strongest interfacial adhesion. It can also be seen from Table 4 that the Wad values of all coherent interfaces are larger than those of all semi-coherent interfaces before and after doping.
3.3 Interface energy and segregation energy
Next, the interface energy of pure and doped interfaces as a function of △μCu is shown in Fig. 5. It is obvious that all the doped interfaces with Sc at S1 site have much smaller γint values compared to Sc at S2 site in the whole range of △μCu considered in this work. The γint values of all the doped interfaces with Sc at S1 site are smaller than those of its corresponding pure interface; however, the γint values of all the doped interfaces with Sc at S2 site are higher than those of its corresponding pure interface, indicating that Sc at S1 site could significantly increase the interface stability, while Sc at S2 site could significantly decrease the interface stability, and the doped interfaces with Sc at S1 site are thermodynamically more stable than Sc at S2 site. In Fig. 5, the γint values of all coherent pure or doped interfaces are smaller than thoseof all semi-coherent interfaces, showing that the coherent interface is more stable than semi-coherentinterface. All in all, the site of Sc has an important influence on interfacial stability, and the influence on coherent and semi-coherent interfaces is significantly different. It can also be seen from Fig. 5 that, before doping, the γint value of pure Cu-rich θ′(001)-Cu/Al(001) interface between the △μCu of -4.087 and-1.2 eV is larger than that of the pure coherent θ′(001)-Cu/Al(001) interface, indicating that the interfacial stability of coherent θ′(001)-Cu/Al(001) interface occupied by interstitial Cu atoms is decreased compared with the interface without interstitial Cu atoms, which is consistent with the results reported in Ref. [38]. While the interfacial stability can be adjusted by changing the chemical potential of Cu. when the △μCu exceeds -1.2 eV, the Cu-rich θ′(001)-Cu/Al(001) interface is more stable than the θ′(001)-Cu/Al(001) interface. After doping, the Cu-rich θ′(001)-Cu/Al(001) interface with bridge site stacking and Sc at S1 site has the smallest γint value, that is, this interface is the most thermodynamically stable, which is in good agreement with the calculation result of the Wad and also shows that Sc doping has a great influence on the interface stablility of Cu-rich θ′(001)-Cu/Al(001) interface.
The total energy and the segregation energy of doped interfaces were calculated, as shown in Fig. 6. It can be seen that all the Eseg values of doped interfaces with Sc at S1 site are negative, and are significantly more negative than the values of doped interfaces with Sc at S2 site, indicating that the ability of Sc at S1 site to segregate at the interface is much stronger than that of at S2 site. It is worth mentioning that the Esegvalue of Cu-rich θ′(001)-Cu/Al(001) interface with bridge site stacking and Sc at S1 site is the most negative, showing that this interface configuration is the most conducive to interfacial bonding, which is in good agreement with the work of adhesion results.
Through the above analysis of interfacial bonding strength, it is revealed that the doped interfaces with Sc at S1 site can significantly decrease the interface energy and increase the work of adhesion. These calculation results are strongly supported by experimental results of GAO et al [56] that Sc atoms prefer to diffuse in the Al site, as well as by previous experiments [29,67] showing that Sc decreases the interface energy and segregates towards the interface.
Table 4 Work of adhesion Wad of pure interfaces and Sc-doped θ′(001)-Cu/Al(001) and θ′(010)/Al(010) interfaces with different terminations and configurations (J/m2)
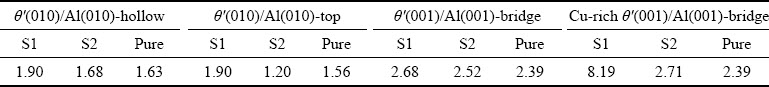
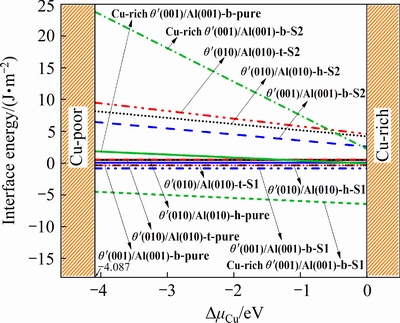
Fig. 5 Interface energies of pure interfacesand Sc-doped θ′(001)-Cu/Al(001) and θ′(010)/Al(010) interfaces with different terminations and configurations (The letters b, t and h in the figure denote bridge, top and hollow sites, respectively, and all the θ′(001)/Al(001) interfaces are Cu-terminated interfaces)
3.4 Bonding characteristics
The bonding properties between atoms at the interface play an important role in the interface bonding strength. In order to further explore the influences of Sc doping on the interfacial strength, the electron localization function (ELF) which characterizes the strength and type of bonds between atoms, the total density of states (TDOS),and the partial density of states (PDOS) were also analyzed.
The ELF value is between 0 and 1, with the upper limit value ELF=1 meaning complete electron localization, the lower limit value ELF=0 meaning complete electron delocalization or no electron, and the middle value ELF=1/2 meaning the electron-gas-like pair probability.
The ELF distributions of doped interfaces and pure interfaces through (111) cutting plane are shown in Fig. 7. Compared with the pure interface, the electron localization at the interface is significantly enhanced at the doped interface in Figs. 7(a-h), which indicates that the bonding interactions between atoms at the interface are significantly strengthened. For the doped coherent interfaces in Figs. 7(a, b) and Figs. 7(e, f), the range of ELF value is 0.6-0.8 between Cu atoms of θ′ slab and Al atoms of Al slab, which indicates that electrons are localized between them and the strong Al-Cu bond is formed. For the doped semi-coherent interfaces in Figs. 7(c, d) and Figs. 7(g, h), the ELF value is around 0.7 between Al atoms of θ′ slab and Al atoms of Al slab, which indicates that the strong Al—Al bond is formed, and a weaker Al—Cu bond also exists. Obviously, the bonding interactions between Al and Cu atoms or Al and Al atoms at the interface with Sc at S1 site are much stronger than those of Sc at S2 site, which also well explains the previous calculation results that the doped interfaces with Sc in S2 site have higher segregation energy, higher interface energy and lower wok of adhesion compared to the interfaces with Sc in S1 site.
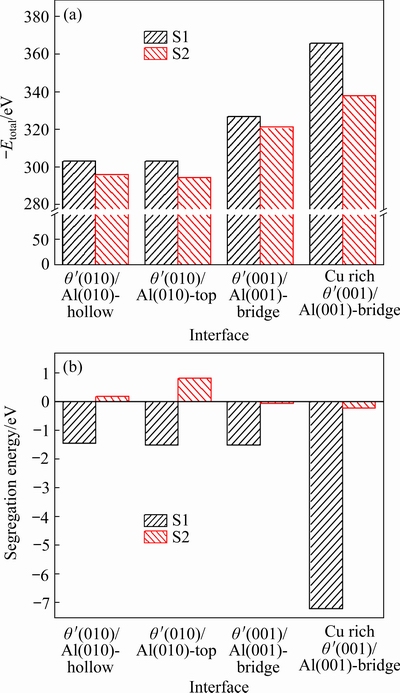
Fig. 6 Total energy (a) and segregation energy (b) of Sc- doped θ′(001)-Cu/Al(001) and θ′(010)/Al(010) interfaces with different terminations and configurations
The TDOS andPDOS of doped interfaces and pure interfaces are shown in Fig. 8. After doping, as shown in Figs. 8(a-h), the orbital hybridization interactions between Al atoms and Cu atoms are more significant in the energy range of -3.5 eV to Fermi energy level, and some overlapping peaks between Al-p and Sc-d orbits appear below the Fermi energy level. Especially the interfaces with Sc at S1 site in Figs. 8(a-d), there is an obvious hybridization between the Al-p orbits and Cu-d orbits, which contributes to the formation of Al—Cu bonds at the interfaces. For the Cu-rich θ′(001)-Cu/Al (001) interface in Fig. 8(b), not only is there a hybridization of Al-p orbits and Cu-d orbits near -1.1, -2.7 and -4.1 eV, but also the great overlapping peaks of Sc-d orbits with Al-s and Cu-d orbits respectively appear near -4.1 eV, indicating that there are strong orbital hybridization interactions at this interface. Combined with the calculation results of ELF, it can be seen that the formation of strong Al—Cu and Al—Al bonds at the doped interfaces contributes to the interface bonding strength.
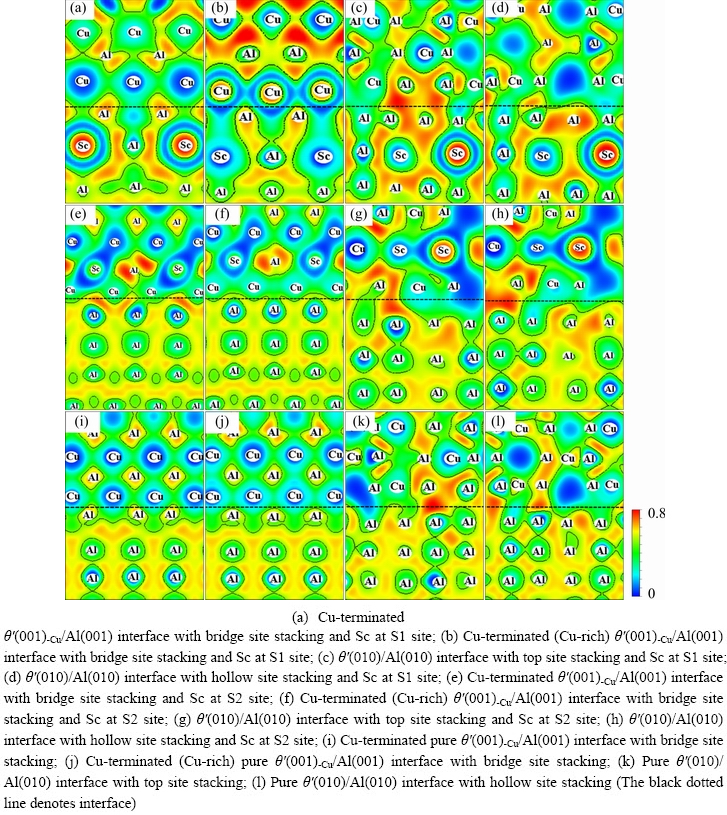
Fig. 7 Electron localization function (ELF) distributions of Sc-doped and pure interfaces
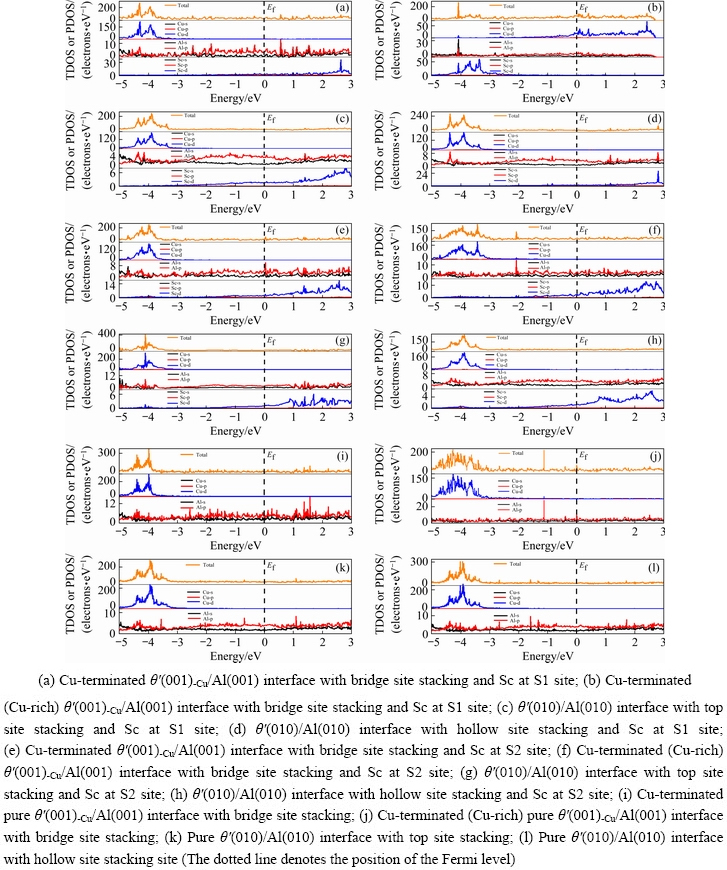
Fig. 8 Total electronic density of states (TDOS)and partial electronic density of states (PDOS) of Sc-doped and pure interfaces
4 Conclusions
(1) The pure coherent Cu-terminated interface with bridge-site stacking is the most stable configuration, while the stability of this interface will decrease when the interstitial Cu occupies the interface.
(2) The doping site of Sc has an important influence on the interface bonding, and Sc atoms have a greater tendency to dope on Al side than onθ′ side.
(3) Sc doping can be effective to improve the bonding strength of the θ′/Al interface, and the formation of strong Al—Cu and Al—Al bonds at the doped interfaces is the main contribution to interface bonding.
(4) The doped coherent Cu-rich θ′(001)-Cu/Al(001) interface with Sc at S1 site has the lowest interface energy and the highest work of adhesion, thus is the most stable interface with strongest bonding strength.
Acknowledgments
Theauthorsaregratefulforthefinancialsupportsfrom theNational Key Research and Development Program of China (No. 2019YFB2006500), and the National Natural Science Foundation of China (Nos. 52171024, 51771234, 51601228). First-principles calculations were partially carried out at the High Performance Computing of Central South University, China.
References
[1] WILLIAMS J C, STARKE E AJr. Progress in structural materials for aerospace systems [J]. ActaMaterialia, 2003, 51(19): 5775-5799.
[2] CHEN Y, WEYLAND M, HUTCHINSON C R. The effect of interrupted aging on the yield strength and uniform elongation of precipitation-hardened Al alloys [J]. ActaMaterialia, 2013, 61(15): 5877-5894.
[3] ZHOU Q, HUA D P, DU Y, REN Y, KUANG W W, XIA Q S, BHARDWAJ V. Atomistic study of atomic structures and dislocation nucleation at Al/Al2Cu interfaces [J]. International Journal of Plasticity, 2019, 120: 115-126.
[4] GUAN Ren-guo, JIN Hong-mei, JIANG Wen-sen, WANG Xiang, WANG Yu-xiang, LI Zheng, ZHANG Jian, LIU Hui-nan. Quantitative contributions of solution atoms, precipitates and deformation to microstructures and properties of Al-Sc-Zr alloys [J]. Transactions of Nonferrous Metals Society of China, 2019, 29(5): 907-918.
[5] HU S Y, BASKES M I, STAN M, CHEN L Q. Atomistic calculations of interfacial energies, nucleus shape and size of θ′ precipitates in Al-Cu alloys [J]. ActaMaterialia, 2006, 54(18): 4699-4707.
[6] LUO Kang, JIANG Yong, YI Dai-qing, FU Shang, ZHANG B. Strained coherent interface energy of the Guinier-Preston II phase in Al-Cu during stress aging [J]. Journal of Materials Science, 2013, 48(22): 7927-7934.
[7] GUINIER A. Interpretation of anomalous X-ray scattering by structurally hardened alloys [J]. ActaCrystallographica, 1952, 5(1): 121-130.
[8] PRESTON G D. Structure of age-hardened aluminium-copper alloys [J]. Nature, 1938, 142(3595): 570.
[9] LIU H, PAPADIMITRIOU I, LIN F X, LLORCA J. Precipitation during high temperature aging of Al-Cu alloys: A multiscale analysis based on first principles calculations [J]. ActaMaterialia, 2019, 167: 121-135.
[10] GUINIER P A. The precipitation mechanism of a metal solid solution crystal: Case of aluminium copper and aluminium silver system [J]. Journal of Physicsand Radium, 1942, 3: 124-136.
[11] KUMAR K S, BROWN S A, PICKENS J R. Microstructural evolution during aging of an AlCuLiAgMgZr alloy [J]. ActaMaterialia, 1996, 44(5): 1899-1915.
[12] SCHNEIDER J, NUNES A C, CHEN P S, STEELE G. TEM study of the FSW nugget in AA2195-T81 [J]. Journal of Materials Science, 2005, 40(16): 4341-4345.
[13] PARK M W, LEE J S, LIM D G, CHOI Y G, KWAK D J, SUNG Y M. Transmission electron microscopy characterization of Al2Cu precipitate growth kinetics by in situ heat treatment [J]. Vacuum, 2008, 83(1): 107-112.
[14] VAITHYANATHAN V, WOLVERTON C, CHEN L Q. Multiscale modeling of θ′ precipitation in Al-Cu binary alloys [J]. ActaMaterialia, 2004, 52(10): 2973-2987.
[15] BOURGEOIS L, DWYER C, WEYLAND M, NIE Jian-feng, MUDDLE B C. Structure and energetics of the coherent interface between the θ′ precipitate phase and aluminium in Al-Cu [J]. ActaMaterialia, 2011, 59(18): 7043-7050.
[16] CHEN Yi-qiang, ZHANG Ze-zhong, CHEN Zhen, TSALANIDIS A, WEYLAND M, FINDALY S, ALLEN L J, LI Jie-hua, MEDHEKAR N V, BOURGEOIS L. The enhanced theta-prime (θ′) precipitation in an Al-Cu alloy with trace Au additions [J]. ActaMaterialia, 2017, 125: 340-350.
[17] MUDDLE B C, RINGER S P, POLMEAR I J. Superconductors, surfaces and superlattices[M].Amsterdam: Elsevier, 1994: 999-1023.
[18] NICHOLSON R B, NUTTING J. Direct observation of the strain field produced by coherent precipitated particles in an age-hardened alloy [J]. The Philosophical Magazine: A Journal of Theoretical Experimental and Applied Physics, 1958, 29(3): 531-535.
[19] GAO Lin, LI Kai, NI Song, DU Yong, SONG Min. The growth mechanisms of θ′ precipitate phase in an Al-Cu alloy during aging treatment [J]. Journal of Materials Science & Technology, 2021, 61: 25-32.
[20] BISWAS A, SIEGEL D J, WOLVERTON C, SEIDMAN D. Precipitates in Al-Cu alloys revisited: Atom-probe tomographic experiments and first-principles calculations of compositional evolution and interfacial segregation [J]. ActaMaterialia, 2011, 59(15): 6187-6204.
[21] BOURGEOIS L, DWYER C, WEYLAND M, NIE Jian-feng, MUDDLE B C. The magic thicknesses of θ′ precipitates in Sn-microalloyed Al-Cu [J]. ActaMaterialia, 2012, 60(2): 633-644.
[22] ROSALIE J M, BOURGEOIS L. Silver segregation to θ′(Al2Cu)-Al interfaces in Al-Cu-Ag alloys [J]. ActaMaterialia, 2012, 60(17): 6033-6041.
[23] FANG H C, CHEN K H, CHEN X, HUANG L P, PENG G S, HUANG B Y. Effect of Zr, Cr and Pr additions on microstructures and properties of ultra-high strength Al-Zn-Mg-Cu alloys [J]. Materials Science and Engineering A, 2011, 528(25-26): 7606-7615.
[24] WANG Ming, HUANG Lan-ping, CHEN Kang-hua, LIU Wen-sheng. Influence of minor combined addition of Cr and Pr on microstructure, mechanical properties and corrosion behaviors of an ultrahigh strength Al-Zn-Mg-Cu-Zr alloy [J]. Micron, 2018, 104: 80-88.
[25] GAO Y H, CAO L F, KUANG J, ZHANG J Y, LIU G, SUN J. Assembling dual precipitates to improve high-temperature resistance of multi-microalloyed Al-Cu alloys [J]. Journal of Alloys and Compounds, 2020, 822: 153629.
[26] SHYAM A, ROY S, SHIN D, POPLAWSKY J D, ALLARD L F, YAMAMOTO Y, MORRIS J R, MAZUMDER B, IDROBO J C, RODRIGUEZ A, WATKINS T R, HANYNES J A. Elevated temperature microstructural stability in cast AlCuMnZr alloys through solute segregation [J]. Materials Science and Engineering A, 2019, 765: 138279.
[27] LIANG S S, WEN S P, WU X L, HUANG H, GAO K Y, NIE Z R. The synergetic effect of Si and Sc on the thermal stability of the precipitates in AlCuMg alloy [J]. Materials Science and Engineering A, 2020, 783: 139319.
[28] YANG C, ZHANG P, SHAO D, WANG R H, GAO L F, ZHANG J Y, LIU G, CHEN B A. The influence of Sc solute partitioning on the microalloying effect and mechanical properties of Al-Cu alloys with minor Sc addition [J]. ActaMaterialia, 2016, 119: 68-79.
[29] GAO Y H, CAO L F, YANG C, ZHANG J Y, LIU G, SUN J. Co-stabilization of θ′-Al2Cu and Al3Sc precipitates in Sc-microalloyed Al-Cu alloy with enhanced creep resistance [J]. Materials Today Nano, 2019, 6: 100035.
[30] ZHAO Wen-yue, SUN Zhi-mei, GONG Sheng-kai. Vacancy mediated alloying strengthening effects on γ/γ′ interface of Ni-based single crystal superalloys: A first-principles study [J]. ActaMaterialia, 2017, 135: 25-34.
[31] WANG Jiong, SHANG Shun-li, WANG Yi, MEI Zhi-gang, LIANG Yong-feng, DU Yong, LIU Zi-kui. First-principles calculations of binary Al compounds: Enthalpies of formation and elastic properties [J]. Calphad, 2011, 35(4): 562-573.
[32] WANG J, DU Y, TAO X, OUYANG Y, ZHANG L, CHN Q, ENGSTR?M A. First-principles generated mechanical property database for multi-component Al alloys: Focusing on Al-rich corner [J]. Journal of Mining and Metallurgy, Section B: Metallurgy, 2017, 53(1): 1-7.
[33] ZHU Long-peng, WANG Jiong, DONG Chen-chen, SHANG Shun-li, DU Yong, LIU Zi-kui. Understanding the surface adsorption and oxidation of cubic Cr0.5Al0.5N by first-principles calculations [J]. Computational Materials Science, 2021, 196: 110518.
[34] ZHU Long-peng, WANG Jiong, DONG Chen-chen, DU Yong, SHANG Shun-li, LIU Zi-kui. Stability, elastic and electronic properties of Ta2N by first-principles calculations [J]. Crystals, 2021, 11(4): 445.
[35] CHONG Xiao-yu, GUAN Pin-wen, HU Ming-yu, JIANG Ye-hua, LI Zu-lai, FENG Jing. Exploring accurate structure, composition and thermophysical properties of η carbides in 17.90wt.%W-4.15wt.%Cr-1.10wt.%V-0.69wt.%C steel [J]. ScriptaMaterialia, 2018, 154: 149-153.
[36] CHONG Xiao-yu, SHANG Shun-li, KRAJEWSKI A M, SHIMANEK J D, DU Wei-hang, WANG Yi, FENG Jing, SHIN D, BEESE A M, LIU Zi-kui. Correlation analysis of materials properties by machine learning: Illustrated with stacking fault energy from first-principles calculations in dilute fcc-based alloys [J]. Journal of Physics: Condensed Matter, 2021, 33(29): 295702.
[37] QIAN Qi, LIU Zheng-qing, JIANG Yong, WANG Yi-ren, AN Xing-long, SONG Min. Basal stacking fault induced twin boundary gliding, twinning disconnection and twin growth in hcp Ti from the first-principles [J]. Transactions of Nonferrous Metals Society of China, 2021, 31(2): 382-890.
[38] KIM K, ZHOU Bi-cheng, WOLVERTON C. Interfacial stability of θ′/Al in Al-Cu alloys [J]. ScriptaMaterialia, 2019, 159: 99-103.
[39] KRESSE G, FURTHM?LLER J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set [J]. Physical Review B, 1996, 54(16): 11169-11186.
[40] WANG Guan-jie, PENG Li-yu, LI Kai-qi, ZHU Liang-gang, ZHOU Jian, MIAO Nai-hua, SUN Zhi-mei. ALKEMIE: An intelligent computational platform for accelerating materials discovery and design [J]. Computational Materials Science, 2021, 186: 110064.
[41] PERDEW J P, BURKE K, WANG Yue. Generalized gradient approximation for the exchange-correlation hole of a many-electron system [J]. Physical Review B, 1998, 54(23): 16533-16539.
[42] FISCHER T H, ALMLOF J. General methods for geometry and wave function optimization [J]. The Journal of Physical Chemistry, 1992, 96(24): 9768-9774.
[43] PFROMMER B G, C?T? M, LOUIE S G, COHEN M L. Relaxation of crystals with the quasi-Newton method [J]. Journal of Computational Physics, 1997, 131(1): 233-240.
[44] CHONG Xiao-yu, HU Ming-yu, WU Peng, SHAN Quan, JIANG Ye-hua, LI Zu-lai, FENG Jing. Tailoring the anisotropic mechanical properties of hexagonal M7X3 (M=Fe, Cr, W, Mo; X=C, B) by multialloying [J]. ActaMaterialia, 2019, 169: 193-208.
[45] MONKHORST H J, PACK J D. Special points for brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188-5192.
[46] LI Ye-fei, GAO Yi-min, XIAO Bing, MIN Ting, FAN Zi-jian, MA Sheng-qiang, YI Da-wei. Theoretical study on the electronic properties and stabilities of low-index surfaces of WC polymorphs [J]. Computational Materials Science, 2011, 50(3): 939-948.
[47] YANG Jian, WANG Yue, HUANG Ji-hua, YE Zheng, SUN Xuan, CHEN Shu-hai, ZHAO Yue. First-principles calculations on Ni/W interfaces in steel/Ni/W hot isostatic pressure diffusion bonding layer [J]. Applied Surface Science, 2019, 475: 906-916.
[48] MUTASA B, FARKAS D. Atomistic structure of high-index surfaces in metals and alloys [J]. Surface Science, 1998, 415(3): 312-319.
[49] SUN Dong-qiang, WANG Yong-xin, LU Yan-li, CHEN Zheng, RAO Qing-lei. Influence of atom termination and stacking sequence on the θ′/Al interfaces from first-principles calculations [J]. Superlattices and Microstructures, 2016, 94: 215-222.
[50] LIU L M, WANG S Q, YE H Q. First-principles study of polar Al/TiN(111) interfaces [J]. ActaMaterialia, 2004, 52(12): 3681-3688.
[51] SIEGEL D J, HECTOR L, ADAMS J B. Adhesion, atomic structure, and bonding at the Al(111)/α-Al2O3(0001) interface: A first principles study [J]. Physical Review B, 2002, 65: 854151.
[52] SIEGEL D J, HECTOR L G, ADAMS J B. First-principles study of metal-carbide/nitride adhesion: Al/VC vs. Al/VN [J]. ActaMaterialia, 2002, 50(3): 619-631.
[53] XIE Hao-nan, ZHAO Nai-qin, SHI Chun-sheng, HE Chun-nian, LIU En-zuo. Effects of active elements on adhesion of the Al2O3/Fe interface: A first principles calculation [J]. Computational Materials Science, 2021, 188: 110226.
[54] HAN Y F, DAI Y B, WANG J, SHU D, SUN B D. First-principles calculations on Al/AlB2 interfaces [J]. Applied Surface Science, 2011, 257(17): 7831-7836.
[55] AARONSON H I, CLARK J B, LAIRD C. Interfacial energy of dislocation and of coherent interphase boundaries [J]. Metal Science Journal, 1968, 2(1): 155-158.
[56] GAO Y H, GUAN P F, SU R, CHEN H W, YANG C, HE C, CAO L F, SONG H, ZHANG J Y, ZHANG X Y, LIU G, NIE J F, SUN J, MA E. Segregation-sandwiched stable interface suffocates nanoprecipitate coarsening to elevate creep resistance [J]. Materials Research Letters, 2020, 8(12): 446-453.
[57] LI Ye-fei, GAO Yi-min, XIAO Bing, MIN Ting, MA Sheng-qiang, YI Da-wei. Theoretical calculations on the adhesion, stability, electronic structure, and bonding of Fe/WC interface [J]. Applied Surface Science, 2011, 257(13): 5671-5678.
[58] TANG Shu-li, GAO Yi-min, LI Ye-fei, ZHENG Qiao-ling. Preparation and interface investigation of Fe/Al2O3P composite activated by Ni and Ti [J]. Advanced Engineering Materials, 2016, 18(11): 1913-1920.
[59] WANG Chan, LIANG Shu-hua, CAO Fei, ZHANG Qiao. Interface microstructure evolution of a novel CuW/Al composite fabricated by an infiltration method [J]. Journal of Alloys and Compounds, 2020, 816: 152506.
[60] CHONG Xiao-yu, PALMA J P S, WANG Yi, SHANG Shun-li, DRYMIOTIS F A, RAVI V E, STAR K, FLEURIAL J, LIU Zi-kui. Thermodynamic properties of the Yb-Sb system predicted from first-principles calculations [J]. ActaMaterialia, 2021: 217.
[61] SIEGEL D J, HECTOR L G, ADAMS J B. Adhesion, stability, and bonding at metal/metal-carbide interfaces: Al/WC [J]. Surface Science, 2002, 498(3): 321-336.
[62] MATSUNAGA K, SASAKI T, SHIBATA N, MIZOGUCHI T, YAMAMOTO T, IKUHARH Y. Bonding nature of metal/oxide incoherent interfaces by first-principles calculations [J]. Physical Review B, 2006, 74(12): 125423.
[63] WITT W. Absolute precision determination of lattice constants on germanium and aluminum single crystals with electron interference [J]. Journal of Natural Research A, 1967, 22(1): 92-95.
[64] KHEIN A, SINGH D J, UMRIGAR C J. All-electron study of gradient corrections to the local-density functional in metallic systems [J]. Phys Rev B, 1995, 51(7): 1-6.
[65] BENALI A, LACAZE-DUFAURE C, MORILLO J. Density functional study of copper segregation in aluminum [J]. Surface Science, 2011, 605(3-4): 341-350.
[66] ESHELMAN F R, SMITH J F. Single-crystal elastic constants of Al2Cu [J]. Journal of Applied Physics, 1978, 49(6): 3284-3288.
[67] GAO Y H, YANG C, ZHANG J Y, GAO L F, LIU G, SUN J, MA E. Stabilizing nanoprecipitates in Al-Cu alloys for creep resistance at 300°C [J]. Materials Research Letters, 2018, 7(1): 18-25.
Al-Cu合金中Sc掺杂θ′/Al界面稳定性和电子结构的第一性原理研究
张冬兰1,汪炯1,孔毅1,邹有2,杜勇1
1. 中南大学 粉末冶金研究院,长沙 410083;
2. 中南大学 信息与网络中心,长沙 410083
摘 要:通过第一性原理计算方法研究Al-Cu合金中Sc掺杂θ′(Al2Cu)/Al的界面特性。根据计算结果和已报道的实验结果,建立Sc掺杂的半共格和共格θ′(Al2Cu)/Al界面(Sc掺杂在Al表面(S1位点),Sc掺杂在θ′表面(S2位点))模型。通过对界面结合强度的分析,发现Sc位于S1位点时,掺杂界面的界面能显著降低,黏附功显著增加。特别是被间隙Cu原子占据的共格界面,当Sc位于S1位点时具有极好的结合强度。电子结构表明,Sc在S1位点的界面形成强Al—Cu键,而Sc在S2位点的界面形成Al—Al键。Al—Cu和Al—Al键的形成对提高掺杂界面强度起着至关重要的作用。
关键词:Al-Cu合金;Sc掺杂θ′/Al界面;界面结合强度;电子结构
(Edited by Wei-ping CHEN)
1003-6326/  2021 The Nonferrous Metals Society of China. Published by Elsevier Ltd & Science Press
2021 The Nonferrous Metals Society of China. Published by Elsevier Ltd & Science Press
Abstract:The properties of Sc-doped θ′ (Al2Cu)/Al interface in Al-Cu alloys were investigated by first-principles calculations. Sc-doped semi-coherent and coherent θ′ (Al2Cu)/Al interfaces (Sc doped in Al slab (S1 site), Sc doped in θ′ slab (S2 site)) were modeled based on calculated results and reported experiments. Through the analysis of interfacial bonding strength, it is revealed that the doping of Sc at S1 site can significantly decrease the interface energy and increase the work of adhesion. In particular, the doped coherent interface with Sc at S1 site which is occupied by interstitial Cu atoms has very good bonding strength. The electronic structure shows the strong Al—Cu bonds at the interfaces with Sc at S1 site, and the Al—Al bonds at the interfaces with Sc at S2 site are formed. The formation of strong Al—Cu and Al—Al bonds plays an important role in the enhancement of doped interface strength.
