
���±�ţ�1004-0609(2010)07-1267-08
Al-Mg-Si�Ͻ���U1��U2���ԭ�ӳɼ�������
��Ӣ�����»�����Τ �ȣ��Ĵ������ƴ���
(������ѧ ������ѧ�빤�̼���ѧԺ������530004)
ժ Ҫ��
Ӧ��EET���ۺĽ���TFD���۶�Al-Mg-Si�Ͻ�ʱЧ������������U1��U2���ԭ�ӳɼ��ͽ���ܽ��м��㡣������������ྦྷ������ǿ���ʹ�ǿ������Al��Si���������Ȼ���Al�����е���ǿ���綼ǿ�öࣻ�������ྦྷ���ж��Խ�ǿ��Al��Si��������Ҫ����Ǽܽṹ������ǿ�������ǿ�ȡ�ǿ���Ͻ�����á�����������U1��������U2���и�ǿ��Al��Si����ṹ���ҽ���ܽϴ���ˣ����U2����˵��U1����ȶ�����������������(001)Al//(110)U1����洦��ɱ��������������ԽϺã�����Ӧ���ܽϵͣ�������ȶ�������(001)Al// (010)U2���������ܶ�ƫ����������������ڴ˽��洦��Ӧ���ϴ���ԭ�Ӽ�ƥ��ϲ���洢��(Ӧ����)�ϸߣ����׳�Ϊ�����κˡ���������������ĵط���
�ؼ��ʣ�
Al-Mg-Si �Ͻ���U1��U2����ԭ�ӳɼ�����ѧ������
��ͼ����ţ�TG111.1���� ���ױ�־�룺A
Atomic bondings and properties of U1 andU2 phases of Al-Mg-Si alloy
GAO Ying-jun, CHEN Hua-ning, WEI Na, WEN Chun-li, HUANG Chuang-gao
(College of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
Abstract: Atomic bondings of U1 and U2 phases in Al-Mg-Si alloy during aging were calculated by using the EET theory and the improved TFD theory. The results show that the strongest bonding and the second strong bonding in two phases are both Al��Si bonds, which are stronger than those in Al matrix. The firm network structures of Al��Si bonds are formed in U1 and U2 phases to enhance the bond network and strengthen alloy, while the bonding networks of Al��Si in U1 phase are not only stronger than those in U2 phase, but also with greater combining energy, therefore the structure of U1 is more stable. The calculation results also show that the electron density on the interface (001)Al//(110)U1 between U1 and matrix of Al is continuous with lower strain energy so that the interface (0001)Al//(110)U1 is more stable, while that on the interface (001)Al//(010)U2 is not continuous with a greater stored energy, poor atom-matching and higher stored energy, which will lead to precipitate a new phase or form a creak to break the alloy.
Key Word: Al-Mg-Si alloy; U1 and U2 phases; atomic bonding; mechanical property
Al-Mg-Si �Ͻ����ھ��е��ܶȡ���ǿ�Ⱥ�������ѧ���ܣ����ѱ��㷺Ӧ���ڳ����ͷɻ��ṹ������ ��[1-2]��ͨ������£�Al������������Mg��Cu�����ı�Ͻ�ʱЧ���̵��������м������࣬����һ��ʱЧ�����£��ӹ�������������������GP�����¡塢�¡�ͦ���[2]������о�[3-5]���֣����й�ʣSi�ģ�l-Mg-Si�Ͻ��ʱЧ�����У������µ�������U1��U2�࣬�Ͻ��ṹ���ݻ�˳�����·�ʽ����[5]��SSSS��(Mg, Si)�Ŵء�GPZ���¡��(�¡�+B��+U1+U2)����(Mg2Si)��U1��U2��һ�������й�ʣSi�ģ�l-Mg-Si �Ͻ��У���ʱЧ�¶�Ϊ200~300 ��ʱ������[4]���úϽ����ṹ�ݻ������в�����������U1��U2�ԺϽ������в�ͬӰ��[5-8]��Ŀǰ����U1��U2��չ���о������࣬��Ҫ����ʵ���϶Ը��ྦྷ��ṹ�IJⶨ�����ȶ��Ե��о����������ϣ��������о��߲��õ�һԭ��������۵��ӽṹ[9-10]�����й����ڲ�ԭ�ӳɼ��ļ��㣬Ŀǰ��û����ر�������ˣ��о���������Щ��������ڲ�ԭ�Ӽ����������Ż��Ͻ���ϵ������зdz���Ҫ�����塣
���ڼۼ�����[11]���ܴ����۽����Ĺ�������Ӿ����������(EET)[12]�Լ��Ľ��Ľ���TFD����[13], �ڴ���������ϵ�Ͻ�ĵ��ӽṹ�����ṩһ�����ʵ�õľ��鷽����ʹ�öԸúϽ������ܵ��о��������뵽ԭ�ӳɼ��ĵ��ӽṹ��Ρ��������߽�EET��Ľ���TFD�������ڶ�Al-Mg-Si�Ͻ�������U1��U2�ڲ�ԭ�Ӽ�ļ۵��ӳɼ���������������γɵĽ�������������м��㣬��ԭ�ӳɼ���������������������ԭ�Ӽ����ص㼰��ԺϽ����ܵ�Ӱ�졣
1 ģ��
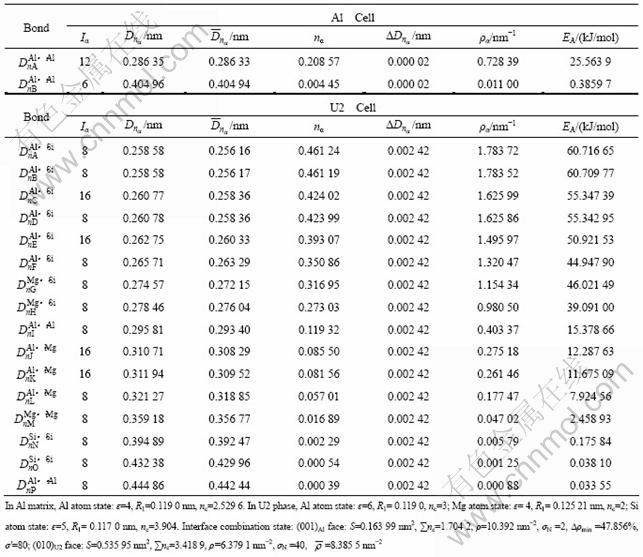
U1��[4]�����Ǿ�ϵ(����Al2CaSi2����La2O3��[14])���ʰ�״����Ϊ50~500 nm����Ϊ50 nm������ʽΪAl2MgSi2����λ��������1��Mgԭ�ӣ�2��Alԭ�ӣ�2��Siԭ�ӣ���������a=b=0.405 1 nm��c=0.674 1 nm��w(Mg)/w(Si)=0.66���ռ�ȺΪ![]() m1(No.164)��U1��ľ����ṹ��ͼ1��ʾ���������Al��ȡ���ϵΪ[7-8]��(001)A1//(110)U1��[310]A1//[001]U1��
m1(No.164)��U1��ľ����ṹ��ͼ1��ʾ���������Al��ȡ���ϵΪ[7-8]��(001)A1//(110)U1��[310]A1//[001]U1��![]()
![]() (001)A1//(110)U1��[100]A1// [0001]U1��
(001)A1//(110)U1��[100]A1// [0001]U1��![]()
![]() ��
��
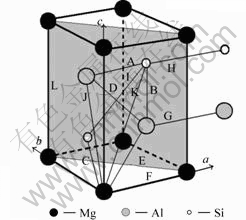
ͼ1 U1�����ṹ
Fig.1 Microstructure of U1 cell
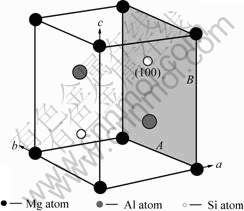
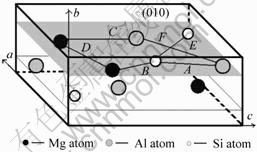
������U2�ṹ[5]����12��ԭ�ӣ����Կ������ɷ���MgAlSi�и�������4��ԭ�Ӷ��õ���(���䵥λ��������4��MgAlSi��Ԫ)��������U2�����ɦ�"������Alԭ�������Mgԭ�Ӻ�Siԭ�ӽ����������ж����ɡ������� U1��U2��������¡��ͦ¡䶼����״���ںϽ�ʱЧ�������γ�ʱ�����Ż���Al�Ĩ�100?����������У�����������ϵ��U2��λ������MgAlSi(Co2Si)��λ����[4, 14]���ƣ��ռ�Ⱥ��Pnma����������a=0.675 2 nm��b=0.405 1 nm��c=0.794 nm��U2��ľ����ṹ��ͼ2��ʾ���������Al��λ���ϵ[5]Ϊ��![]()
![]() [130]Al// [001]U2��
[130]Al// [001]U2��
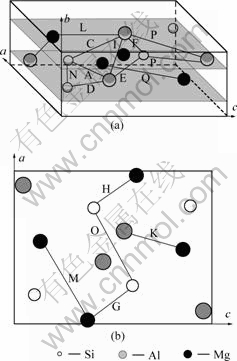
ͼ2 U2�����ṹ
Fig.2 Microstructure of U2 cell: (a) Bond network structure in U2 cell; (b) Bond network structure along [010]
2 ��������
������ԭ�ӵļ۵��ӽṹ�ڱ�����ָ�ù�����ԭ��������״̬�Լ�ԭ���γɹ��ۼ��ļ���ֲ�������EET[12-13]���ۣ�ԭ�ӵĹ��۵����Ƿֲ�����������ڡ��ν����Լ�s����ԭ�ӵļ��ϵġ������Ϲ��۵��Ӷ���(������na)������ԭ�Ӽ��ʽ��ʾ��
![]() (1)
(1)
ʽ�У�Duv�ǹ��ۼ��ࣻRu��Rv�ǵ�����ࣻ��Ϊ�������µ���ֵ������[11-12]�еĹ�ʽȷ����u��v�ֱ��ʾuԭ�Ӻ�vԭ�ӣ�n����ʾu��vԭ���γɵļ��������ڵĹ��۵����������������̹�ϵ��
![]() (2)
(2)
ʽ�У�k1��k2�ֱ�Ϊ������ u��vԭ�ӵĸ�����![]() ��
��![]() �ֱ�Ϊu��vԭ�ӵĹ��۵�������I��Ϊn�������ĵ�ͬ����������ͬ������ѡȡ����������[15]�����ķ�����ȷ����
�ֱ�Ϊu��vԭ�ӵĹ��۵�������I��Ϊn�������ĵ�ͬ����������ͬ������ѡȡ����������[15]�����ķ�����ȷ����
���ڸ������Ľṹ��ȷ����ʵ�龧�������� ��[4-5]��������ˣ����ü����(BLD)����[12]������ǿ��nA���̣����μ�����[15-18]����ⲽ�裬����ʽ(1)��(2)����������������ԭ�ӳɼ��ļ۵��ӽṹ, ������BLD�о�ȷ��ԭ�ӵ��ӽ�״̬�š�����õ��ĸ������Ĺ��ۼ�������1��4��ʾ������ncΪ���۵�����������Ϊ��λ�����ĵ����ܶȣ�EAΪ������ܣ�?��Ϊ�����ܶȲ![]() Ϊƽ�������ܶȣ�SΪ����ο���Ԫ���������Ϊ��������������ԭ��״̬��Ŀ���ҡ�Ϊ���ֲ�����������ԭ��״̬��Ŀ��
Ϊƽ�������ܶȣ�SΪ����ο���Ԫ���������Ϊ��������������ԭ��״̬��Ŀ���ҡ�Ϊ���ֲ�����������ԭ��״̬��Ŀ��
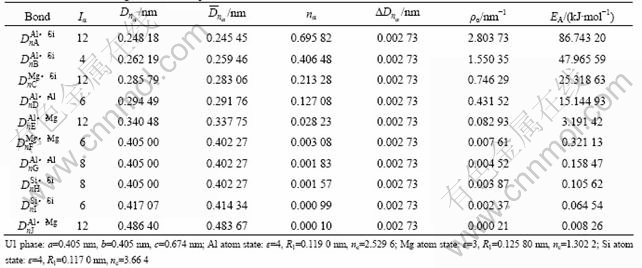
��1 ����������U1��ԭ�ӳɼ�
Table 1 Atomic bonding of metastable phase U1
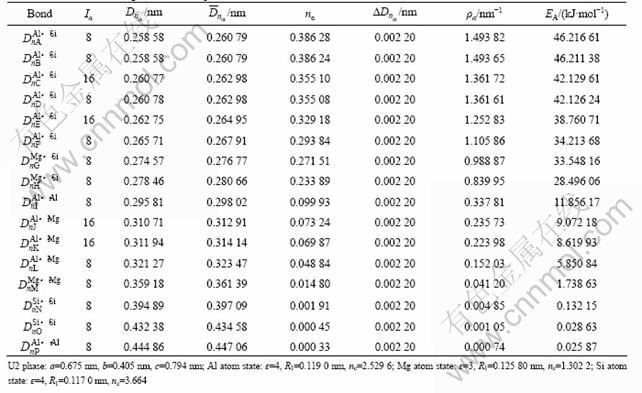
��2 ������U2��ԭ�ӳɼ�
Table 2 Atomic bonding of metastable phase U2
��3 (001)Al // (110)U1������ԭ�ӳɼ�
Table 3 Atomic bonding of (001)Al // (110)U1 interphase boundary

����[13]���������������ӽṹ��ָ�����������ӽṹ���������������ƽ���ϵļ�����ӷֲ��⣬���������������ƽ���ϵ�ƽ�����۵����ܶȦ�(hkl)����(uvw)�������ܶȵ���Բ�ֵ?�Ѻ�ʹ��������ܶ���һ�������±���������ԭ��״̬������(��һ�������£���?�ѣ�10%���жϽ�������ܶȵ������ԡ���?�ѣ�10%ʱ���ѵ����ܶȶ���Ϊ�����������ԽϺã���?�ѣ�10%ʱ���ѽ�������ܶȶ���Ϊƫ�������������Խϲ�)������洦�����ܶȦ����ߣ������ԭ�Ӽ����Խ�ܣ������ϵþ�Խ�ι̣�����洦�ĵ����ܶȲ���С�������ϵĵ����ܶ������Ծ����ã�����ԭ�Ӽ���ƥ��þ�Խ�ã���������ܾ�Խ�ͣ��������Ӧ��Ҳ��С����֮���������Ӧ������������ܾ�Խ�ߣ������Խ���ȶ���������Ӧ����һ��ֵʱ�������ܶȵ���������ƻ�����������������ɻ��ں���ϳ��ֶ��ѡ������ܶ������Եĺû�ʵ�����ǵ���ԭ�Ӽ����������µĽ����ֱ��Ӱ�쵽���ϵ����ܡ�
�����������ƽ��(hkl)��(uvw)�ϵ�ƽ�����۵����ܶȦ�(hkl)�ͦ�(uvw)�����洦�����ܶȵ���Բ�ֵ?�ѣ��ɷֱ����ݸĽ���TFD����[13]�������¹�ʽ��
![]() ,
,![]() (3)
(3)
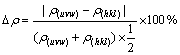 (4)
(4)
ʽ�У�![]() ��nAIA+nBIB+nCIC+����Ϊ��ο���Ԫ�ϵĹ��۵���������S(uvw)��S(hkl)�ֱ�Ϊ(uvw)��(hkl)��ο���Ԫ�������
��nAIA+nBIB+nCIC+����Ϊ��ο���Ԫ�ϵĹ��۵���������S(uvw)��S(hkl)�ֱ�Ϊ(uvw)��(hkl)��ο���Ԫ�������
2.3.1 U1��U2����ڲ�ԭ�Ӽ�ǿ
�ڼ���U1��U2��ļ����ʱ�����������о�|?Duv(na)|��0.005 nm�Ľ���ж���⡣����BLD�����жԶ��Ĵ�������[12, 16]�����ڵ��������Ҫ�ų���ȫ����������ӵ��ӽ����(��Al��6, ��Mg��4, ��Si��6)����������[16]�ķ�����һ������£�����������ͽ����仯������Alԭ�Ӵ�ദ�ڵ�3��4��5�ӽף����ڵ�4��5�ӽ��������Mg�ǵ��ͽ��������������ɵ��ӵĵ�4�ӽĿ����Ժ�С������Mg�Ļ�е���ܾ�����Mg����һ���Ľ��ǿ�Ⱥͽ���ܣ��� Mgԭ�Ӵ��ڵ�1��2�ӽ�ʱ���۵���̫�٣������ϻ�е���ܵ�Ҫ����ˣ������Mg�Ľ����������е����Ҫ���ԭ��״̬Ӧ���ڵ�3�ӽס�����Si��������Ժ�С�����ۼ���Ϻ�ǿ����һ����Ҳ����Siԭ��ͨ�����ڼ۵��Ӷ�Ϊ���۵��ӵĵ�6�ӽס����ܺϽ���ԭ�Ӽ������ö�ԭ���ӽ�Ǩ����һ��Ӱ�죬ʹ�úϽ���ԭ�Ӳ���һ���ʹ�������ܵ��ӽף�������Ҳ˵����ͨ��ԭ�Ӽ����������£��ӽ�Ǩ��һ��������ܵ��ӽ����仯����Mgԭ��״̬��ʱҲ������ڵ�2��4�ӽס�ͬ����Siԭ��״̬��ʱҲ������ڵ�4��5�ӽס�
��4 (001)Al// (010)U2������ԭ�ӳɼ�
Table 4 Atomic bonding of (001)Al// (010)U2 interphase boundary
ԭ���ӽ�״̬��ȷ�����˸������Ϲ����⣬��ʱ���迼�Ǿ���Ľ���ܣ�������������[19]�����ļ������ܹ�ʽ�õ�����ֵ���ٸ��ݽ���ܼ���ֵ��ʵ�鴦��ֵ[14]����ϵij̶����жϽ���Ƿ��������������[19]�ļ��㹫ʽ������Ѽ���õ���ԭ�Ӽ�ǿ��������о�����õ�U1��U2�Ľ������ʵ��ֵ���Ǻϵúܺã�����С��10��(U1��Ľ���ܣ� ����ֵEU1��-2.0327��103 kJ/mol�����ʵ��ֵ[9, 20]E��U1= -1.7151��103 kJ/mol��U2��Ľ���ܣ�����ֵEU2= -4.073��103 kJ/mol�����ʵ��ֵ[9, 20]E��U2=-3.6967��103 kJ/mol)�����õ�һԭ������õ��Ľ����[9]��ʵ��ֵͨ������15%��
2.3.2 U1��U2��Ľ���ԭ�Ӽ�ǿ
��������[13]����Ľ�����ӽṹ���ۣ����������۵��ӽṹ��������ռ�۵��ӽṹ�����ϣ����ǵ���Ҫ������ռ��еļ�����о�|?Duv(na)|��0.005 nm��ͬʱ��Ҫ����������������������������ܶȲ�?����С�����������մ�ԭ������(001)Al// (110)U1��(001)Al// (010)U2�����ĵ��ӽṹ���õ��Ľ�����3��4���С�
ͼ3 U1����(100)��ļ��ṹ
Fig.3 Bonding structure of plane (100) in U1 cell
ͼ4 U2����(010)��ļ��ṹ
Fig.4 Bonding structure of plane (100) in U2 cell
3 ����
EET��������[13]�ж������ṹ���Ӱ������¼���������������N ��nA ����A ��EA�����У���NΪ��ṹ�п��ܴ��ڵ�ԭ��״̬��������N������ṹ���ȶ�����ṹ����nAΪ�����ȶ�״̬(���Ǧ�N����ܵ�ԭ����̬����Ϊ���ܵ�ԭ����̬����ǿ���ϵ�nAֵ)ʱ��ǿ���ۼ��ϵĹ��õ��Ӷ������������������ԭ�������ɵĹ��ۼ���ǿ����nAֵ�����ۼ���ǿ����֮����������Ȼ��nAֵ�Ĵ�С��һ���̶��ϱ���������߽���ع������׳̶ȣ���nA����������ļ����ɢ���ع������ѡ���ṹ���Ӧ�AΪ��ǿ���ۼ��ϵĵ�λ�����ĵ����ܶ�(����A��nA/DA)��������������nA���ơ���ṹ����EAΪ������ܣ���������£�
![]() (5)
(5)
ʽ�У�![]() ΪA���ϵ�����ԭ�ӵ��ۺϵ�����γ�������ֵ�ϵ�����ɸýṹ��ԭ�ӵĵ��ӶԺ˵�ɵ���������ϵ��bu��bv�ļ�Ȩƽ��ֵ��
ΪA���ϵ�����ԭ�ӵ��ۺϵ�����γ�������ֵ�ϵ�����ɸýṹ��ԭ�ӵĵ��ӶԺ˵�ɵ���������ϵ��bu��bv�ļ�Ȩƽ��ֵ��![]() Ϊ�ṹ��Ԫ������ԭ�Ӽ���ۺϳɼ���������ֵ�ϵ��ڽṹ��Ԫ�и�ԭ�ӳɼ������ļ�Ȩƽ��ֵ��n��/DnAΪ�ýṹ�е���ǿ���ۼ��ϵļ������ܶȣ���ֵ��
Ϊ�ṹ��Ԫ������ԭ�Ӽ���ۺϳɼ���������ֵ�ϵ��ڽṹ��Ԫ�и�ԭ�ӳɼ������ļ�Ȩƽ��ֵ��n��/DnAΪ�ýṹ�е���ǿ���ۼ��ϵļ������ܶȣ���ֵ��![]() ��
��![]() ����������[12]�����IJ�����á�
����������[12]�����IJ�����á�
�ɱ�1��4�ӽṹ������Կ�����������U1��U2��Alԭ�Ӻ�Mgԭ��״̬�ֱ�ȡ�봿Al��Mg������ԭ�Ӵ���ͬ����״̬[16]����Alԭ�Ӵ��ڵ�4�ӽף�Mgԭ�Ӵ��ڵ�3�ӽס���Siԭ����ȡ���ڽϵ͵ĵ�4�ӽ�(��Si�����д��ڵ�6�ӽף��������)���乲�۵����ɴ�Si������4���ٵ�3.664�������������0���ӵ�0.336��
�ɱ�1��4���ɼ���������U1��U2����ǿ��ΪAl��Si������ǿ��ΪAl��Si�������ϵĹ��۵�����nA��nB�����ܶȦ�A����B�Լ�����EA��EB���Ȼ������Al����ǿ���ļ�ǿ(��nA(Al)=0.208 57������=0.728 39 nm-1��EA=25.563 9 kJ/mol)Ҫ��2~3�������У�U2���м���õ���Ҫ��Al��Si���ļ�ǿ�Ȼ������Al����ǿ���ļ�ǿ��ǿ���ɴ˿ɼ���������U1��U2���ھ��н�ǿ��ԭ�Ӽ��磬�������п�����ǿ�Ͻ�������ǿ��
��1��2����ΪEET����Ľṹ���ӵļ�����������U1�࣬nA=0.695 82����A=2.803 73 nm-1��EA= 86.743 20 kJ/mol������U2�࣬nA=0.386 28����A=1.493 82 nm-1��EA=46.216 61 kJ/mol����Ȼ��U1�����ǿ���ļ�ǿ��U2��Ľ�������������U1��U2���EA(U1)= 86.743 2 kJ/mol��EA(U2)��46.216 6 kJ/mol(EA(U1)��EA(U2))��˵��Al��Si��������γ�U1�࣬��������[9]�������������������������������U1���U2�࣬��ǿ���ʹ�ǿ������Al��Si�������ΪMg��Si�������ң�Al��Si����������Mg��Si��Al��Mg��Al��Al��Mg��Mg��Si��Si����ǿ�ܶ࣬˵��U1��U2�����γ�����Al��Si��Ϊ���ļ���Ǽܽṹ����ͼ5��ʾ������ܼ�����������EU1=-2.032 7��103 kJ/mol��EU2=-4.073��103 kJ/mol������U2���U1������ȶ��Ը��á�
����EET��������[13]�����������ӵ���������Ϊ?�ѣ�Ϊ����洦�ĵ����ܶȲ��Ϊʹ��������ܶȱ���������ԭ��״̬���������ҡ�Ϊƫ������(��?�ѣ�10%)�����Ŀ���ԭ��״̬������������Ϊ�����ϵĵ����ܶ�(��)����������ǿ��������洦�ĵ����ܶȲ�?�ѣ�10%ʱ��?����С�������ϵĵ����ܶ����������ã��������Ӧ����С��ʹ�����ܶȱ��������Ŀ���ԭ��״̬���������࣬������Խ����Խ�ȶ�����֮���������Ӧ�������������Խ�ߣ�����������ȶ���������Ӧ���ﵽһ��ֵʱ�������ܶ���������ƻ�����ʱ��������������ɻ��ں���ϳ��ֶ��ѡ�

ͼ5 U1��U2������ļ���
Fig.5 Bonding networks of U1 (a) and U2 (b) phases
�ɱ�3��4�ɼ���(001)Al//(110)U1����洦ƽ��������ܶȦ�U1=11.906 9 nm-2����(001)Al//(010)U2����洦��ƽ��������ܶȦ�U2=8.385 5 nm-2Ҫ��˵������(001)Al//(110)U1��Ͻ�ǿ��U1���洦�ĵ����ܶȲ�?��min=0.082 4%(��10%)����U1������ɱ��������������ԽϺã���˵��U1�������������Ӧ����С������ԭ�Ӽ�ƥ��ýϺã�������ȶ�������Ӧ���ܽϵͣ������ڻ���ľ�����������(001)Al// (010)U2���������ܶ�?��min=47.856%(��10%)��ƫ��������������ˣ��ڴ˽��洦Ӧ���ϴ���ԭ�Ӽ�ƥ��ýϲ���洢��(Ӧ����)�ϸߣ���ʱ�������׳�Ϊ�����κ���������������������ĵط���
4 ����
1) ����ռ�۵��ӽṹ�У�������U1��U2�е����ԭ��Al��Mg�������봿Ԫ�ؾ����е�ԭ����ͬ���ӽף���Al�����ڵ�4�ӽף�Mg�����ڵ�3�ӽף���Siԭ��״̬�����ı䣬Ǩ�Ƶ���4�ӽ�״̬��
2) ������U1��U2����ǿ����ΪAl��Si������ǿ��ΪAl��Si�������Ȼ���Al�����е���ǿ��Ҫǿ2~3����˵��U1��U2�����������ǿ�Ͻ�������ǿ��������U1����ǿ���ۼ���������U2����ǿ���ۼ�Ҫǿ��2�������ǿ��Ҳ��U2�����ǿ��ǿ������U1���Alԭ����Siԭ�Ӿ��и�ǿ�Ľ����������ˣ�U1���U2�������������
3) (001)Al// (110)U1����洦��ɱ��������������ԽϺã�����Ӧ���ܽϵͣ�������ȶ�������(001)Al// (010)U2���������ܶ�ƫ��������������ˣ��ڴ˽��洦Ӧ���ϴ���ԭ�Ӽ�ƥ��ýϲ���洢�ܽϸߣ����׳�Ϊ�����κˡ���������������ĵط���
REFERENCES
[1] HIROSAWA S, SATO T. Nano-scale clusters formed in the early state of phase decomposition[J]. Mater Sci Forum, 2005, 475/479: 357-360.
[2] FUKUI K, TAKEDA M. The metastable phase responsible for peak hardness and its morphology in an Al-Mg-Si alloy[J]. Mater Sci Forum, 2005, 475/479: 361-364.
[3] VISSERS R, van HUIS M A, ZANDBERGEN H W, MARIOARA C D. The crystal structure of the �£�phase in Al-Mg-Si alloys[J]. Acta Mater, 2007, 55: 3815-3823.
[4] ANDERS G F, RAGNVALD H. Bonding in MgSi and Al-Mg-Si compounds relevant to Al-Mg-Si alloys[J]. Phys Rev B, 2003, 67: 224106.
[5] ANDERSEN S J, MARIOARA C D, FR?SETH A, VISSERS R, ZANDBERGEN H W. Crystal structure of the orthorhombic U2 precipitate in the Al-Mg-Si alloy system and its relation to the �£�and ��" phases[J]. Mater Sci Eng A, 2005, 390: 127-138.
[6] TSAO C S, CHEN C Y. Precipitation kinetics and transformation of metastable phases in Al-Mg-Si alloys[J]. Acta Mater, 2006, 54: 4621-4631.
[7] ANDERSEN S J, MARIOARA C D, VISSERS R, FROSETH A, ZANDBERGEN H W. The structural relation between precipitates in Al-Mg-Si alloys[J]. Mater Sci Eng A, 2007, 444: 157-169.
[8] MATSUDA K, SAKAGUCHI Y, MIYATA Y. Precipitation sequence of various kinds of metastable phases in Al-Mg-Si alloy[J]. J Mater Sci, 2000, 35: 179-189.
[9] RAVI C, WOLVERTON C. First-principles study of crystal structure and stability of Al-Mg-Si-(Cu) precipitates[J]. Acta Mater, 2004, 52: 4213-4227.
[10] VAN M A, CHEN J H. Phase stability and structural relations of nanometer-sized, matrix-embedded precipitate phases in Al-Mg-Si alloys in the late stages of evolution[J]. Acta Mater, 2006, 54: 2945-2955.
[11] ����L. ��ѧ������[M]. ¬����, ����. �Ϻ�: �Ϻ���ѧ����������, 1996: 393.
PAULING L. The nature of chemical bonds[M]. LU Jia-xi, et al, transl. Shanghai: Shanghai Science and Technology Press, 1996: 393.
[12] ������. ��������Ӿ����������[M]. ����: ���ֿ�ѧ����������,1993.
ZHANG Rui-lin. Empirical electron theory in solids and molecules[M]. Changchun: Jilin Science and Technology Press, 1993.
[13] ��־��, ��־��, ��ΰ��. ������ӽṹ���������[M]. ����: ��ѧ������, 2002: 23.
LIU Zhi-lin, LI Zhi-lin, LIU Wei-dong. Electron structure and properties of interface[M]. Beijing: Science Press, 2002: 23.
[14] GREGORY A L, ROALD H. The TiNiSi family of compounds: Structure and bonding[J]. Inorg Chem, 1998, 37: 5754-5763.
[15] GAO Ying-jun, HUANG Chuang-gao, HOU Xian-hua. Atomic bonding and property of Al-Mg-Sc alloy[J]. Materials Transaction, 2005, 46: 1148-1153.
[16] �����. ����þ��Ԫ����ͼ��, �����Լ���-Al12Mg17��ļ۵��ӽṹ����[J]. ���ִ�ѧѧ��, 1979, 4(4): 54-66.
YU Rui-huang. Analysis of valence electron structure about ��, �� phase and ��-Al12Mg17 phase in Al-Mg binary phase diagram[J]. Journal of Jilin University, 1979, 4(4): 54-66.
[17] GAO Ying-jun, HOU Xian-hua, MO Qi-feng. Atomic bonding of precipitate and phase transformation of Al-Cu-Mg alloy[J]. J Alloy & Compounds, 2007, 441: 241-245.
[18] GAO Ying-jun, MO Qi-feng, CHEN Hua-ning. Atomic bonding and mechanical properties of Al-Li-Zr alloy [J]. Mater Sci Eng A, 2009, 499: 299-303.
[19] ����, ������, �����. ���ɽ��������ᄃ�����ܼ���[J]. �й���ѧ A, 1988, 18(3): 323-327.
XU Wan-dong, ZHANG Rui-lin, YU Rui-huang. Calculation of crystal binding energy in transition metal compounds[J]. Science in China (Series A), 1988, 18(3): 323-327.
[20] ������һ, ��. ����ѧ��������[M]. ������, ����. ����: ��ѧ������, 1979: 85.
Shuichi Iida , et al. Mathematical tables of physics[M]. ZHANG Zhi-xian, et al, transl. Beijing: Science Press, 1979: 85.
(�༭ �� ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(50661001, 50061001); ����ʡ��Ȼ��ѧ����������Ŀ(0991026, 0832029, 0639004)
�ո����ڣ�2009-10-09�������ڣ�2010-01-22
ͨ�����ߣ���Ӣ��������; �绰: 0771-3232666; E-mail: gaoyj@gxu.edu.cn
