
��������������Һ��Cu�ĵ��ӽṹ����������
�ջԽ�, л����, ���콨����£�����棬������
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ������ ��ɳ��410083)
ժҪ������SGTE���ݿ�ľ����ȶ�������Debye-Gr��neisenģ�ͣ����ô�������ԭ��(OA) �����о�����������������Һ��Cu��ԭ��״̬��ԭ�����ܡ�ԭ�Ӷ��ܡ�ԭ��������嵯��ģ����������ϵ���������������¶ȵı仯��ϵ���о�������������ӽṹ����������õ�һԭ���������õĽ��һ�£������뾶��ԭ�Ӷ��ܺ�ԭ���������¶����������ӣ���������ϵ������ֵ��ʵ��ֵ���Ǻϣ���Һ���ʱ��Liquid-Cu�����ɵ��Ӻ��۵��Ӿ���Ǽ�����ת�ƣ�����d������s����ת�ƣ����ӽṹ��ת�併��Һ��ĵ����ԣ�����Һ��ԭ����λ�ķ����ԣ�����ԭ�ӵ����뾶���������
�ؼ��ʣ�
FCC-Cu��Liquid-Cu�����ӽṹ��Debye-Gr��neisenģ����CALPHAD������
��ͼ����ţ�TG111 ���ױ�ʶ�룺A ���±�ţ�1672-7207(2007)01-0001-08
Electronic structures and physical properties of FCC
and metastable liquid Cu metals
TAO Hui-jin, XIE You-qing, PENG Hong-jian, YU Fang-xin, LIU Rui-feng, LI Xiao-bo
(School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: Combining the lattice stability parameters of SGTE database with Debye-Gr��neisen model, the atom states, atomic potential and vibrating energies, atomic volumes, bulk moduli and linear thermal expansion coefficients of FCC and metastable Liquid Cu metals in SGTE database of pure elements were studied using one-atom(OA) theory. The results show that calculated electronic structures agree well with those of first principles; the single bond radius, atomic vibrating energy and atomic potential energy increase with the increase of temperature; the linear thermal expansion coefficients agree well with the experimental data as well; and when FCC-Cu is transformed into Liquid-Cu, the free and covalent electrons are transferred into nonbonding electrons, the covalent electrons in d orbital are transferred into s orbital and this change of electronic structures leads to the decrease of electrical conductivity, the weakening of direction of atom bonding, the increase of the single bond radius and the atomic volumes of Liquid-Cu.
Key words: FCC-Cu��Liquid-Cu��electronic structure��Debye-Gr��neisen model; CALPHAD method
��ͼ�ļ���ģ��(CALPHAD)��ָ���²��Ͽ�����Ƶ�ǿ��������[1-2]���Դ����ʲ�ͬ����ṹ���Gibbs�ܼ������ȶ�������������CALPHAD����Ҫ������Ϊ�ˣ���Ҫ�Դ�����Ԫ����ѧ��ϵ����ʵ�����������������������������һ����[3]�����У�SGTE(Scientific Group Thermodata Europe) ���������ݿ�[4]������298.15 K����78��Ԫ�صIJ�ͬ����ṹ��Gibbs�ܱ���ʽ����Ӧ�ľ����ȶ���������һԭ�����ƺ����Ͳ��������к�����ƣ��ڲ�����ʵ�����ϵ�����¿���ȷ�������ʲ�ͬ����ṹ������ȶ��ԡ�Y.Wang��[5]�õ�һԭ������ϵͳ�о���78�ִ�����FCC, HCP �� BCC�ṹ������ȶ��ԣ��������CALPHAD�������õĽ�����жԱȣ����ֶ��ߴ��ڽϴ���[5]���Ͻ�ϵͳ��ѧ���(Systematic sciences of alloys, SSA)[6]�Ľ�������Ϊ�˼�С�����������ֲ��̽���Դ�������Ͻ���ӽṹ������ѧ���ʺ��������ʵ�ͳһ������SSA����Զ�ԭ������� ��[7-8]������������[9]��ԭ��״̬�ӻ�����[10-11]�������������ʵ�������Ϊ��������Ag-Cu[12-15]��Ti-Al[16-18]��Au-Cu[19-21]�ȺϽ�ϵ��ԭ��״̬��ԭ�����ܡ�ԭ�Ӷ��ܡ�ԭ��������嵯��ģ����������ϵ���Ȳ������¶Ⱥͳɷֵı仯���ɽ����о�����SSA����У�������Ĺ����ǶԸ��ֲ�ͬ����ṹ�����ʵ�״̬���������¶ȵı仯��ϵ���м���ģ�⣬��ԭ��(One atom, OA)����������һ���������ۻ������ڴˣ��������߽�CALPHAD�����ľ����ȶ�������Debye-Gr��neisenģ�����ϣ�����OA����ȷ���µ����ۼ�����·����������һ��·������������������Һ��Cu�ĵ��ӽṹ���Լ����ǵ������������¶ȵı仯��ϵ��
1��ԭ���뷽��
1.1����������ԭ������
1.1.1��ԭ��״̬����
���ȶ�������ԭ����ȣ�����̬�����е�ԭ����ͨ����ѧ����ϵġ���ˣ�����ԭ�����ĵ��ӿ����չ��ܷ�Ϊ�ɼ��ͷdzɼ�������[22]����������һ�ְ������۵��ӡ����ɵ��ӻ���Ե��ӵĻ�ϼ�����ˣ�ԭ�����ɼ��ļ۵��ӿ��Է�Ϊ���۵��ӡ����ɵ��Ӻʹŵ��ӣ��dzɼ�����(��ƷǼ�����)����Ϊ���л�ѧ���Ե�����ʵ���ӡ����У����۵��ӶԽ��������Ҫ���ã����ɵ��ӶԵ��硢���Ⱥ���������Ҫ���ã����ŵ��ӶԲ��ϴ�������Ҫ���á�
��OA�����У��û���ԭ��̬��k(k=1,2,��,n)�����ӻ�(�����)���õ�������ռ����(Quasi-Electron-Occupation ,QEO)������ԭ��״̬�ף�
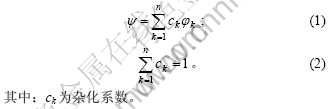
ÿһ������̬��k����ѭPauli
������ԭ������Խ���Cu, ![]() ��
��![]() ��
��![]() �ֱ��ʾ��k������ԭ��̬��s��p��d ����Ĺ��۵�������
�ֱ��ʾ��k������ԭ��̬��s��p��d ����Ĺ��۵�������![]() Ϊ���ɵ�s̬��������
Ϊ���ɵ�s̬��������![]() Ϊ�Ǽ���d��������
Ϊ�Ǽ���d��������![]() (��λΪ0.1 nm)Ϊ�ѸĽ���Pauling�����뾶[23]��nc��nf��nv�ֱ��ʾ�ܵĹ��۵��ӣ����ɵ��Ӻͳɼ��������������
(��λΪ0.1 nm)Ϊ�ѸĽ���Pauling�����뾶[23]��nc��nf��nv�ֱ��ʾ�ܵĹ��۵��ӣ����ɵ��Ӻͳɼ��������������![]() ��дΪ�ƣ���Cu�ĵ�ԭ��״̬�������������¹�ʽ�����
��дΪ�ƣ���Cu�ĵ�ԭ��״̬�������������¹�ʽ�����

1.1.2������������
�����±�s��ȡֵ1��2�ȱ�ʾ����ڡ��ν��ڵȹ��ۼ���GsΪ�ɾ���ṹ���;����ij�����RΪ�����뾶����ΪPauling�ļ�����[23]��ncΪ�ܵĹ��۵�������IsΪ��s���ڼ��ĵ�ͬ��������������Ϊ��

1.1.3������ܺ��ƺ���
��ԭ�������(Many-atom- interactions, MAI)���ܺ�����������ʽ���
 ����
����
1.1.4���嵯��ģ������������ϵ��
�����嵯��ģ���Ķ���![]() ��������ѹǿ�Ĺ�ϵ
��������ѹǿ�Ĺ�ϵ![]() ���Եõ���
���Եõ���
![]()
������¶ȵı仯���Ը���Debye-Gr��neisenģ��[24]ȷ������������ϵ����ȷ����

1.2���������
1.2.1������ԭ��̬
Eckardt[25]ͨ���о����֣����ڴ���ǿ�ҵ�s-p�ӻ�������Cu���δռ�ݵ�p�ܼ�ʵ�����Ѿ���Ч��s�ܼ�����ˣ�p�ܼ��ĵ��ӿ�����Ϊs̬���ӡ��������ֽ��Ʒ��������ļ���10�ֻ���ԭ��̬����Ӧ��FCC�ṹ����ľ������ͽ���ܣ�������1��ʾ�����У�aΪ��������EcΪճ���ܡ�
��1������Cu�Ļ���ԭ��̬����ӦFCC�ṹ����ľ������ͽ����
Table 1��Lattice constants and cohesive energies ofFCC-Cu in basic atom states
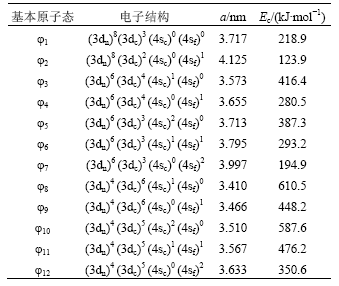
1.2.2��������·
����ԭ��̬ȷ��֮��OA������CALPHADȷ�������ȶ������ļ��㷽�����ϣ���������Debye-Gr��neisenģ�Ϳ��Լ�����������������Һ��Cu��ԭ��״̬�������������¶ȵı仯��������·��ͼ1��ʾ�������Ǻš�*����ʾʵ�����ݡ����°��ռ������ֲ�������ϸ˵����
��1��������Pauling�����뾶![]() �뾧����a���¶ȷ���ͬ�����ı仯����Debye�¶�*��D[26]�����¶ȱ仯������298.15 Kʱ������FCC-Cu��*
�뾧����a���¶ȷ���ͬ�����ı仯����Debye�¶�*��D[26]�����¶ȱ仯������298.15 Kʱ������FCC-Cu��*![]() [24]�� *��D��R����OA���ۺ�Debye-Gr��neisenģ�Ϳ��Լ���FCC-Cu��0 Kʱ�ľ������͵����뾶��
[24]�� *��D��R����OA���ۺ�Debye-Gr��neisenģ�Ϳ��Լ���FCC-Cu��0 Kʱ�ľ������͵����뾶��
��2������0 Kʱ���Ծ�����a=0.360 3 nm�ͽ����*Ec=336 kJ?mol-1 [26]Ϊ�оݣ��Ա�1�еĻ���ԭ��̬����ɸѡ�����֦�4����9�ͦ�12��̬��������Ͽ����ھ���Ҫ��֮���ҵ�ͬʱ���Ͼ������ͽ���ܱ���ԭ��״̬�ף��ɽ����Ec�õ�ԭ�����ܦ�p���ٸ������ܺ�������嵯��ģ��B�����õ�0 KʱFCC-Cu��Debye �¶ȡ��������������뾶������ܡ����ӽṹ��ԭ�����ܺ��嵯��ģ������*��D��a��R��*Ec���ף���p��B�ȡ�
![]()
![]()
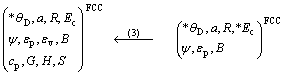
![]()
![]()
![]()

ͼ1��ȷ��FCC-Cu��Liquid-Cuԭ��״̬���������ʵļ�����·ͼ
Fig.1��Schematic procedure for determining atomic states and properties of FCC and Liquid pure Cu-metals
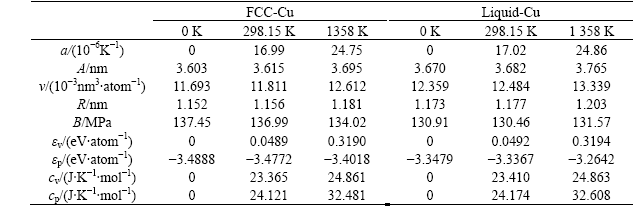
��3���������¶����ߣ�FCC-Cu�ļ۵��ӽṹ����û�б仯�����ԣ����һԭ������ѧ����������[27-28]��ʵ�ʼ�������0 Kʱ�ĵ��ӽṹ��Ϊ�۵����������¶ȵĵ��ӽṹ�����ǣ���Ϊ����ԭ��״̬����һ���������������뾶�������������ͣ������������ԭ��״̬���¶ȵı仯ȡ���ڵ����뾶���¶ȵı仯��ͨ������õ�*��D��a��R��Ec���ף���p����v��B��cp��G��H��S�Ȳ������¶ȱ仯����������Ц�v��cp��G��H��S�ֱ����ԭ�����ܡ���ѹ���ݡ�Gibbs�ܡ��ʺ��ص�����ѧ������
��4������298.15 Kʱ������CALPHAD����ȷ���ľ����ȶ�������GLiquid-FCC[4]�õ�Liquid-Cu��Gibbs�ܣ�
![]()
��Ϊ��һ������Gibbs�ܲο�̬��ѡ���أ����ԣ�(10)ʽͬ����������0 Kʱ�Ļ�̬����ԭ��Ϊ�ο�̬���������ʱ����H(0 K)=-Ec(0 K)[29]�����ң�
![]() �� (11)
�� (11)
�����������[10]�еĽ��ƹ�ʽ������Liquid-Cu��Debye�¶ȣ�
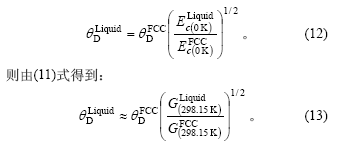
��5����Ϊ�˶�Liquid-Cu�����ʽ��н���ģ�⣬��������Ȼ���ֶ̳������FCC�ṹ[30]������ԭ���������������[31]�ĵ�Ч������裬����ԭ�������ȫ��ͬ�������£�������Liquid-Cu��Ϊ��Ч��FCC���壬��Gr��neisen�����еij���k ��Q0��FCC-Cu
�ṹ����ͬ������![]() ��
��![]() ������Debye-Gr��neisenģ�Ϳ��Լ���Liquid-Cu 0 Kʱ�Ľ����
������Debye-Gr��neisenģ�Ϳ��Լ���Liquid-Cu 0 Kʱ�Ľ����![]() ��ͬʱ�����ݽ���Cu���۵�ʱ���ܶ�[32]�õ�
��ͬʱ�����ݽ���Cu���۵�ʱ���ܶ�[32]�õ�
ƽ��ԭ���������ת��Ϊ��Ч�ľ���������ģ�ͼ���õ�0 Kʱ�ľ�����a�����ջ��Liquid-Cu��0 Kʱ����Ϣ(��D��a��Ec)��
��6�������2�����ƣ���0 Kʱ������Liquid-Cu�ľ�����a=0.367 0 nm�ͽ����Ec =323 kJ?mol-1���Ա�1�еĻ���ԭ��̬����ɸѡ�����ֶԦ�1����5�ͦ�12������̬�ӻ������ҵ�ͬʱ���㾧�����ͽ���ܾ���Ҫ���ԭ��״̬�ף�Ȼ���ɽ����Ec�õ�ԭ�����ܦ�p�����ܺ������Ӷ�����嵯��ģ��B������֪(��D��a��R��Ec���ף���p��B)�Ȳ�����
��7�������3�����ƣ������¶����ߣ�Liquid-Cu�ĵ��ӽṹ�ײ��䣬�����뾶�뾧����ͬ���������ͣ������ۺ�ģ�ͼ���ͬ���õ�(��D��a��R��Ec���ף���p��B��cp��G��H��S)��ϵ�в�����
2������
2.1��ԭ��״̬
ͼ2��ʾΪFCC-Cu��Liquid-Cu�Ծ������ͽ����Ϊ�������ӻ����������ҵ���a�ߺ͵�Ec�ߵĽ��㣬�����ݽ����Ӧ����̬�ӻ��ɷ�ȷ����ԭ��״̬�ɷ�ͼ�����ӻ��ɷּ���2��
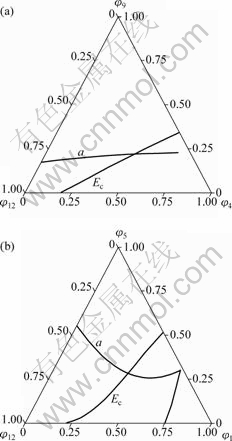
(a) FCC-Cu; (b) Liquid-Cu
ͼ2��FCC-Cu��Liquid-Cu��̬�ӻ��ɷ�ͼ
Fig.2��Composition positions of atomic states of FCCand Liquid pure Cu-metals
��2��FCC-Cu��Liquid-Cu��̬�ӻ��ɷֺ�����
Table 2��Composition percents and properties of FCCand Liquid pure Cu-metals at 0 K

���������ӻ��ɷ�ȷ��FCC-Cu��Liquid-Cu��ԭ��״̬���ó�����ӽṹ�͵����뾶Ϊ
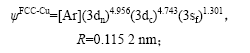
![]()
![]()
2.2����������
2.2.1�����ܺ���
��nx��mx����MAI�ƺ���Wx(r)�е�ָ����r0Ϊƽ��״̬�����ԭ�Ӽ��࣬EcΪ����ܣ���FCC-Cu��Liquid-Cu��Wx(r)�����3��ͼ3��ʾ��
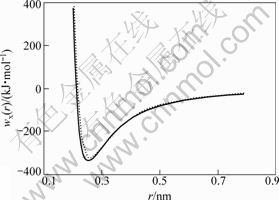
ͼ3��FCC-Cu��Liquid-Cu��Wx(r)��������
Fig.3��The potential energy curves of FCC and Liquid pure Cu-metals
��3��FCC-Cu��Liquid-Cu��Wx(r)�ƺ�������
Table 3��Parameters of Wx(r) potential function of FCC and Liquid Cu-metals

2.2.2����������ϵ�����嵯��ģ��
Debye-Gr��neisenģ�Ͳ������4��ʾ�����У���������ϵ���е�ʵ��������������[24]��ͼ5��������������ϵ������������ԭ������������뾶���嵯��ģ�����¶ȵı仯��ϵ�������ƺ����ɵõ�ͼ4��ʾ���嵯��ģ�����¶ȵı仯���ߡ�
��4��FCC-Cu��Liquid-Cu��Debye-Gr��neisenģ�ͼ������
Table 4��Constants of Debye-Gr��neisen model for FCCand Liquid pure Cu-metals

2.2.3��ԭ�Ӷ��ܺ�����
ԭ�Ӷ��ܺ��������¶ȵı仯��ϵ��ͼ5��Ϊ�˸��õض����Ƚϣ���0 K, 298.15 K���۵�ʱ����ز������жԱȣ��������5��
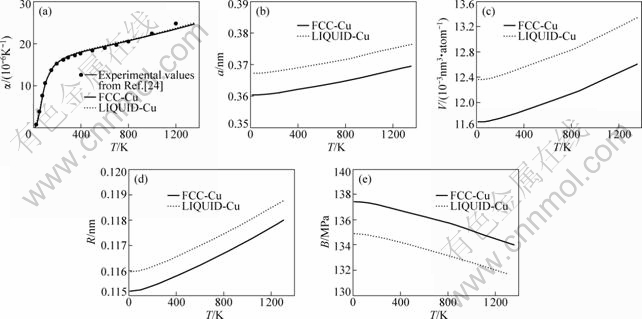
(a) ��������ϵ����(b) ��������(c) ԭ�������(d) �����뾶��(e) �嵯��ģ��
ͼ4��FCC-Cu��Liquid-Cu����������ϵ������������ԭ������������뾶���嵯��ģ�����¶ȵı仯
Fig.4��Relationship between temperature dependence and linear thermal expansion coefficients, lattice constants,
atomic volumes, single bond radiuses and bulk moduli of FCC-Cu and Liquid-Cu
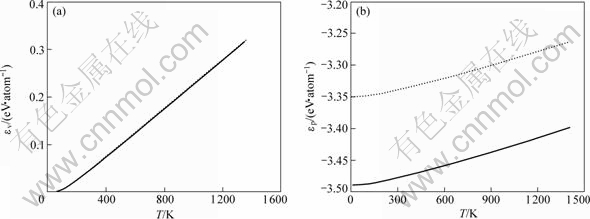
(a) ԭ�Ӷ��ܣ�(b) ԭ������
ͼ5��FCC-Cu��Liquid-Cu��ԭ�Ӷ��ܺ��������¶ȵı仯
Fig.5��Temperature dependence of atomic vibration energy and potential energy of FCC-Cu and Liquid-Cu
��5��FCC-Cu��Liquid-Cu��0K��298.15 K���۵�ʱ���������ʶԱ�
Table 5��Properties of FCC and Liquid pure Cu-metals at 0 K, 298.15 K and 1 358 K
3������������
3.1��FCC-Cu�ĵ��ӷֲ���
������OA���۷������õ��ӽṹ����õ�һԭ����������Ԫ����LRC [25]���ü��������жԱȣ��������6��
��6����ͬ�����о�FCC-Cu���õ��ӷֲ����ĶԱ�
Table 6��Atomic populations of FCC-Cu by various methods
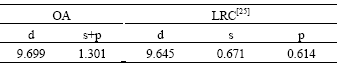
�ɱ�6��֪������OA���۷������õĽ������õ�һԭ�����õĽ��һ�£����⣬������OA���۷������ü��������һԭ��VASP����[5]�Լ����������ƺ���[33]���ü��������жԱȣ��������7��
��7����ͬ�����о�FCC-Cu�������������ĶԱ�
Table 7��Calculated properties of FCC-Cu using different methods
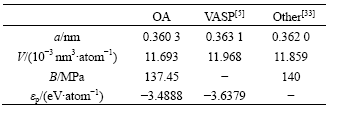
�ӱ�7��֪������OA���۷������������������ɾ����ƺ������ý�����ӽ�������õ�һԭ�����õĽ�����ϴ�ͬʱ����ͼ5�ͱ�5��֪�����ļ������������ϵ�����¶ȵı仯��ϵ��ʵ�������Ǻϣ���������ԭ������͵����뾶���¶ȷ���ͬ���������ͣ��嵯��ģ�����¶��������½������½����Ȳ�����ͼ6�ͱ�5֪��ԭ�Ӷ��ܺ�ԭ�����ܾ����¶����߶����������Ҷ������¶����ӷ������������¶����ӷ��ȵ�3~4�������¶����������У���������յ�������Ҫ��������ԭ�ӵĶ��ܡ�
3.2��Liquid-Cu
��SGTE���������ݿ����۵����µ�����Һ��Cu����ԭ��������������µĵ�Ч������裬�����ּ�����Ч�Եļ�����Ҫͨ����ʵ�����ݵĶԱ���˵��������ʵ������֪������Cu�۵����ϵ�Һ�����ݺ�����۵������[34]�����ҵ�����Һ�����۵�ת��Ϊ�ȶ�Һ��ʱ��������䣬���ǵĺ�ѹ����Cp��ȣ����ԣ����ļ����Liquid-Cu�۵�ʱ�����ݼ�Ϊ�ȶ�Һ������ݡ���8��ʾΪ�۵��¶�ʱFCC-Cu��Liquid-Cu�����ݼ���ֵ��SGTE���ݿ���������ʵ��ֵ�������Աȡ��ɼ������ļ���ֵ��ʵ��ֵ������С���ر���Һ��������ʵ�����ݽ��Ǻϣ��Ӷ�֤��������������Ч�ġ�
��8��1 358 KʱFCC-Cu��Liquid-Cu�����ݼ���ֵ��SGTE���ݿ�����ʵ�����ݵıȽ�
Table 8��Isobaric heat capacity of FCC and Liquid Cu at 1 358 K

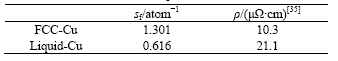
�Ե��ӽṹ���жԱȷ������֣�Liquid-Cu�����ɵ�����0.616��FCC-Cu��1.301�����˽�1�룬��Һ��絼���½������������ķ���2.04[35]һ�¡���9��ʾΪ�۵��¶�ʱ�����ʵ�ʵ������
��9���۵�ʱFCC-Cu��Liquid-Cu�����ɵ�����������ʵıȽ�
Table 9��Comparison between the number of free electrons and electrical resistivity at melting point of FCC
and Liquid Cu
�Աȹ�Һ����ӽṹ�����֣�������Һ��ת��ʱ��ԭ�����������ɵ��Ӻ�ԭ�Ӽ�Ĺ��۵��Ӿ���Ǽ�����ת�ƣ�������ǿ�Ĺ���d��������ԳƵĹ���s����ת�ƣ��Ӷ�������ԭ��֮����λ�ķ����ԣ�������ԭ��֮����λ�������ԡ����ӽṹ��ת��ʹLiquid-Cu�����뾶��ʽ(3)�еĹ���d�������ܼ۵������еı�����С������ԭ�ӵ����뾶��ԭ����� ����
4���ᡡ��
a. �ںϽ�ϵͳ��ѧ(SSA)����£����ô�������ԭ��(OA)���۶�FCC-Cu��ԭ��״̬��ԭ�����ܡ�ԭ�Ӷ��ܡ�ԭ��������嵯��ģ����������ϵ���������������¶ȵı仯�����о������ֵ��ӽṹ�ļ���������õ�һԭ�����ý��һ�£������뾶���¶����߷������ͣ���������ϵ����ʵ��ֵ���Ǻϣ��嵯��ģ�����¶������������½���ԭ�Ӷ������¶����ӵķ������������¶����ӷ��ȵ�3~4������ϵ���յ�������Ҫ��������ԭ�Ӷ��ܡ�
b. ����ԭ���������ĵ�Ч������裬��SGTE���������ݿ����۵����µ�����Һ��Cu����OA�����о������ֲ�������ģ�ⷽ���õ����ȶ�Һ��ĺ�ѹ������ʵ��ֵ�����С����ʵ�������Ǻϡ�
c. ��FCC-Cu��Liquid-Cuԭ��״̬��OA���۽����о����֣�������Һ��ת��ʱ��ԭ�����������ɵ��Ӻ�ԭ�Ӽ�Ĺ��۵��Ӿ���Ǽ�����ת�ƣ�ͬʱ������ǿ�Ĺ���d��������ԳƵĹ���s����ת�ƣ����ӽṹ��ת�併��Һ��ĵ����ԣ�����Һ��ԭ����λ�ķ����ԣ����¹���d�������ܼ۵������еı�����С������ԭ�ӵ����뾶���������
�ο����ף�
[1] Kaufman L, Bernstein H. Computer calculation of phase diagram[M]. New York: Academic Press Inc, 1970.
[2] Saunders N, Miodownik A P. CALPHAD (Calculation of Phase��Diagrams): A comprehensive guide[M]. New York: Pergamon, 1998.
[3] Saunders N, Miodownik A P, Dinsdale A T. Metastable lattice stabilities for the elements[J]. Calphad-Computer Coupling of Phase Diagrams and Thermochemistry, 1988, 12: 351-374.
[4] Dinsdale A T. SGTE data for pure elements[J]. CALPHAD, 1991, 15(4): 317-425.
[5] Wang Y, Curtarolo S, Jiang C, et al. Ab initio lattice stability in comparison with CALPHAD lattice stability[J]. CALPHAD, 2004, 28: 79-90.
[6] XIE You-qing, TAO Hui-jin, PENG Hong-jian, et al. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC TiAl2 type alloys[J]. Physica B, 2005, 366: 17-37.
[7] XIE You-qing. A new potential function with many-atom interactions in solid[J]. Science in China: Series E, 1993, 36(1): 90-99.
[8] XIE You-qing. Relationship of Lennard-jones potential and Morse potential with Wx(r) potential[J]. Transactions of Nonferrous Metals Society of China, 1994(4): 63-66.
[9] XIE You-qing, ZHANG Xiao-dong, ZHAO Li-yin, et al. Electronic structure and properties of Cu metal[J]. Science in China: Series A, 1993, 36(4): 487-494.
[10] XIE You-qing, MA Liu-yin, ZHANG Xiao-dong, et al. Microstructure and properties of Cu-Ni alloys[J]. Science in China: Series A, 1993, 36(5): 612-623.
[11] XIE You-qing. Electronic structure and properties of pure iron[J]. Acta Metallurgica Materialia, 1994, 42(11): 3705-3715.
[12] XIE You-qing. Atomic energies and gibbs energy functions for Ag-Cu alloys[J]. Science in China: Series E, 1998, 41(2): 146-156.
[13] XIE You-qing, ZHANG Xiao-dong. Atomic volumes and volume functions for Ag-Cu alloys[J]. Science in China: Series E, 1998, 41(2): 157-168.
[14] XIE You-qing, ZHANG Xiao-dong. Electronic structure of Ag-Cu alloys[J]. Science in China: Series E, 1998, 41(3): 225-236.
[15] XIE You-qing, ZHANG Xiao-dong. Phase diagram and thermodynamic properties of Ag-Cu alloys[J]. Science in China: Series E, 1998, 41(4): 348-356.
[16] XIE You-qing, PENG Kun, LIU Xin-bi. Influences of xTi/xAl on atomic states, lattice constants and potential-energy planes of ordered FCC TiAl-type alloys[J]. Physica B, 2004, 344: 5-20.
[17] XIE You-qing, LIU Xin-bi, PENG Kun, et al. Atomic states, potential energies, volumes, stability, and brittleness of ordered FCC TiAl3-type alloys[J]. Physica B, 2004, 353: 15-33.
[18] XIE You-qing, PENG Hong-jian, LIU Xin-bi, et al. Atomic states, potential energies, volumes, stability and brittleness of ordered FCC Ti3Al-type alloys[J]. Physica B, 2004, 362: 1-17.
[19] YU Fang-xin, XIE You-qing, NIE Yao-zhuang. Electronic structure of Au-Cu alloys[J]. Transactions of the Nonferrous Metals Society of China, 2004, 14(6): 1041-1049.
[20] л����. Au-Cu�Ͻ�ϵ�������������ľ�����[J]. ����ѧ��, 1998, 34(12): 1233-1242.
XIE You-qing. Lattice constants of disordered and ordered phases in the Au-Cu system[J]. Acta Metallurgica Sinica, 1998, 34(12): 1233-1242.
[21] л����, ������. Au-Cu�Ͻ���۽ṹ������[J]. ����ѧ��: A��, 1994, 30(12): 531-539.
XIE You-qing, ZHANG Xiao-dong. Microstructure and properties of Au-Cu alloys[J]. Acta Metallurgica Sinica, 1994, 30(12): 531-539.
[22] GUO Yi-qing, YU Rui-huan, ZHANG Rui-lin, et al. Calculation of magnetic properties and analysis of valence electronic structures of LaT13-xAlx(T=Fe, Co) compounds[J]. Journal of Physical Chemistry B, 1998, 102(1): 9-16.
[23] Pauling L. Nature of the chemical bond[M]. Ithaca: Cornell University Press, 1960.
[24] Kirby R K, Hahn T A, Rothroch B D. Thermal expansion[C]// Gray D E. American Institute of Physics Handbook. New York: McGraw-Hill Book Company, 1972: 119-138.
[25] Eckardt H, Fritsche L, Noffke J. Self-consistent relativistic band structure of the noble metals[J]. J Phys F: Met Phys, 1984, 14: 97-112.
[26] Kittel C. Solid state physics[M]. New York: John Wiley and Sons Inc, 1976.
[27] Ozolin V, Wolverton S C, Zunger A. Cu-Au, Ag-Au, Cu-Ag, and Ni-Au intermetallics: First-principles study of temperature- composition phase diagrams and structures[J]. Phys Rev B, 1998, 57(11): 6427-6443.
[28] Wei S H, Mbaye A A, Ferreira L G, et al. First-principles calculations of the diagrams of noble metals: Cu-Au, Cu-Ag and Ag-Cu[J]. Phys Rev B, 1987, 36(8): 4163-4185.
[29] �¾���, ��л�. ������Ͻ��еĹ�̬���[M]. ����: ұ��ҵ������, 1997: 8-10.
CHEN Jing-rong, LI Cheng-ji. Phase transitions of solids in metals and alloys[M]. Beijing: Metallurgical Industry Press, 1997: 8-10.
[30] XIE You-qing, DENG Yong-ping, LIU Xin-bi. Electronic structure and physical properties of Cr, Mo, W metal[J]. Transactions of Nonferrous Metals Society of China, 2003, 13(5): 1102-1107.
[31] Barkonyi I, Elbert H, Liechtenstein A I. Electronic structure and magnetic susceptibility of the different structural modifications of Ti, Zr and Hf metals[J]. Physcal Review B, 1993, 48: 7841-7849.
[32] Weast R C. CRC handbook of chemistry and physics, 70th ed[M]. Florida: CRC Press Inc, 1990: B-216.
[33] Zhang Z J. Calculation of the properties of some metals and alloys[J]. Journal of Physics: Condens Matter, 1998, 10: L495-L499.
[34] Chase M W. NIST-JANAF thermochemical tables fourth edition part I[M]. Gaithersburg: National Institute of Standards and Technology, 1998.
[35] Cusack N E. The electronic properties of liquid metals[J]. Reports on Progress in Physics, 1963, 26(1): 361-409.
�ո����ڣ�2006-05-15
������Ŀ��������Ȼ��ѧ����������Ŀ(No.50271085, 50471058)
����飺�ջԽ�(1976-)���У����������ˣ���ʿ�о��������¼�������о�
ͨѶ���ߣ��ջԽ����У���ʿ�о������绰��0731-8879287(O)��E-mail: taohuijin@hotmail.com
[4] Dinsdale A T. SGTE data for pure elements[J]. CALPHAD, 1991, 15(4): 317-425.
[20] л����. Au-Cu�Ͻ�ϵ�������������ľ�����[J]. ����ѧ��, 1998, 34(12): 1233-1242.
[21] л����, ������. Au-Cu�Ͻ���۽ṹ������[J]. ����ѧ��: A��, 1994, 30(12): 531-539.
[23] Pauling L. Nature of the chemical bond[M]. Ithaca: Cornell University Press, 1960.
[26] Kittel C. Solid state physics[M]. New York: John Wiley and Sons Inc, 1976.
[29] �¾���, ��л�. ������Ͻ��еĹ�̬���[M]. ����: ұ��ҵ������, 1997: 8-10.
