

First principles study on electronic structure and optical properties of novel Na-hP4 high pressure phase
SHI Yi-min(ʩ����)1, YE Shao-long(Ҷ����)2
1. Department of Basic Teaching, Hunan Institute of Technology, Hengyang 421000, China;
2. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China
Received 19 November 2009; accepted 3 April 2010
Abstract:
The electronic structure and optical properties of novel Na-hP4 high pressure phase at different pressures (260, 320, 400 and 600 GPa) were investigated by the density functional theory (DFT) with the generalized gradient approximation (GGA) for the exchange and correlation energy. The band structure along the higher symmetry axes in the Brillouin zone, the density of states (DOS) and the partial density of states (PDOS) were presented. The band gap increases and the energy band expands to some extent with the pressure increasing. The dielectric function, reflectivity, energy-loss function, optical absorption coefficient, optical conductivity, refractive index and extinction coefficient were calculated for discussing the optical properties of Na-hP4 high pressure phase at different pressures.
Key words:
first principles; novel Na-hP4 phase; high pressure phase; density functional theory; electronic structure; optical properties;
1 Introduction
At ambient conditions, the alkali metal sodium is free-electron-like crystal with simple structure and behaviors. However, under higher pressures, it is not a simple metal no longer. It was predicted by first- principle calculations and confirmed by experiments [1-2] that sodium undergoes a sequence of symmetry breaking transitions driven by the Peierls mechanism. HANFLAND et al[2] and MCMAHON et al[3] showed that sodium transforms first from the BCC to FCC phase at 65 GPa, and then to a more complex cubic structure, ![]() at 103 GPa. This
at 103 GPa. This ![]() structure was widely reported as a broken-symmetry structure[4-5]. The recent X-ray single-crystal synchrotron diffraction studies of sodium as a function of pressure and temperature (up to 130 GPa at 500 K and up to 150 GPa at room temperature)[6] indicated that sodium exhibits six different structures in this region. In the last significative experimental and theoretical study, MA et al[7] reported experimental observations of a pressure-induced transformation of Na into an optically transparent phase at about 200 GPa, and their experimental and computational data identified the new phase as a wide bandgap dielectric with a six-coordinated, highly distorted double-hexagonal close packed structure along the c-axis (Na-hP4). They attributed the emergence of this dense insulating state to p�Cd hybridizations of valence electrons and their repulsion by core electrons into the lattice interstices[7-8]. To the best of our knowledge, there are no reports of optical properties of this high pressure phase. In this work, the electronic structure and optical properties of Na-hP4 high pressure phase were investigated at 260, 320, 400 and 600 GPa, respectively, by the first principles.
structure was widely reported as a broken-symmetry structure[4-5]. The recent X-ray single-crystal synchrotron diffraction studies of sodium as a function of pressure and temperature (up to 130 GPa at 500 K and up to 150 GPa at room temperature)[6] indicated that sodium exhibits six different structures in this region. In the last significative experimental and theoretical study, MA et al[7] reported experimental observations of a pressure-induced transformation of Na into an optically transparent phase at about 200 GPa, and their experimental and computational data identified the new phase as a wide bandgap dielectric with a six-coordinated, highly distorted double-hexagonal close packed structure along the c-axis (Na-hP4). They attributed the emergence of this dense insulating state to p�Cd hybridizations of valence electrons and their repulsion by core electrons into the lattice interstices[7-8]. To the best of our knowledge, there are no reports of optical properties of this high pressure phase. In this work, the electronic structure and optical properties of Na-hP4 high pressure phase were investigated at 260, 320, 400 and 600 GPa, respectively, by the first principles.
2 Calculation method and model
The calculations are based on a total-energy pseudopotential plane-wave method[9] within the generalized gradient approximation (GGA) in the scheme of Perdew-Burke-Eruzerhof (PBE)[10-11]. The ion-electron interaction is modeled by ultra-soft local pseudo-potentials in the Vanderbilt form[12]. The maximum plane-wave cutoff energy is taken as 410 eV. The geometrical optimization is made and the convergence is assumed when the forces on atoms are less than 10 meV/?. Based on this, the electronic structure is calculated. And the Monkhors-Pack form method[13] is used, which has high symmetrical k points, to calculate the Brillouin zone integration with k grid to be 10��10��6 and do all the energy calculation in the reciprocal space. Finally, within the electric-dipole approximation, the imaginary parts of the dielectric functions ��2(��) can be calculated from the momentum matrix elements between the occupied and unoccupied wave functions within the selection rules, and the real part of dielectric function ��1(��) can be evaluated from the imaginary part of dielectric function by Kramer- Kr?nig relationship[14-16]. Therefore, we can calculate and analyze all solid macroscopical optical constant, such as reflectivity R(��), energy-loss function L(��), optical absorption coefficient I(��), optical conductivity ��(��), refractive index n(��) and extinction coefficient k(��), by using the imaginary part of dielectric function ��2(��) and the real part of dielectric function ��1(��). All the calculations are done by using CASTEP code[16].
The crystal structure of Na-hP4 is space group P63/mmc (No.194), and the sketch of Na-hP4 is presented in Fig.1. There are two inequivalent atomic positions: Na1 at 2a (0, 0, 0) and Na2 at 2d (2/3, 1/3, 1/4). The experimental lattice parameters at 320 GPa are (the cell volume is 0.006 50 nm3 per atom): a=b=0.278 4 nm and c=0.387 3 nm[7]. The equivalent hydrostatics pressure is used in optimization. After optimization, the lattice constants are a=b=0.278 8 nm, and c=0.386 6 nm. The theoretical cell parameters after optimization are similar with the experimental results and the errors are less than 0.181%. The value of c/a is 1.391 and the error is 0.324% compared with the experimental value [7]. The other theoretical cell parameters at different pressures after optimization are listed in Table 1.
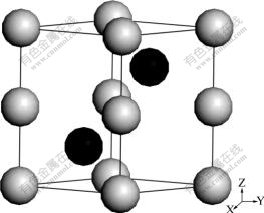
Fig.1 Sketch of Na-hP4 high pressure phase (Gray balls stand for Na1-2a (0, 0, 0) and black balls stand for Na2-2d (2/3, 1/3, 1/4))
Table 1 Calculated lattice parameters of Na-hP4 phase at different pressures
The electronic band structure of Na-hP4 at different high pressures from 260 GPa to 600 GPa along the high- symmetry directions of the Brillouin zone are shown in Fig.2. It is seen that Na-hP4 phase at high pressure is transparent with the valence band maximum (VBM) and conduction band minimum (CBM) at the gamma (��) point. The direct band gaps are about 1.258, 1.808, 2.473 and 3.932 eV at 260, 320, 400 and 600 GPa respectively. The band gap increases and the energy band expands to some extent with increasing the pressure. The results of band gaps at different pressures in present work are similar with the results in Ref.[7] calculated with DFT-PW and DFT-PAW methods, while are greatly underestimated compared with the GW method[7].
The partial density of states (PDOS) of Na1, Na2 and Na of Na-hP4 phase at 320 GPa are presented in Fig.3. It can be seen that below the Fermi level there are three sub-bands. The lowest-lying valence band from -60.8 eV to -58.3 eV is of Na 1s state character, while the valence band from -58.3 eV to -56.4 eV is of Na 2s state character. The middle-lying valence band from -34.5 eV to -30.3 eV consists of Na 1p state and Na 2p state with predominant Na 1p state character, while the valence band from -30.3 eV to -27.6 eV is predominantly of Na 2p state character. The top valence band from -7.4 eV to -3.0 consists of Na 1s state and Na 2s state, and the valence band from -3.0 eV to Fermi level consists of Na 1s state, Na 2s state and Na 2p state with predominant Na 2p state character. The lowest-lying conductive band up to 5.0 eV is a mixture of Na 1s, p states and Na 2s, p states, and mainly consists of Na 1s state and Na 2s state, while the up conductive band is a mixture of Na 1p state and Na 2p state with predominant Na 1p state character. The PDOS of Na1, Na2 and Na of Na-hP4 phase at other high pressure values have the similar character.
Figs.4-9 show the optical functions of Na-hP4 phase at 260, 320, 400 and 600 GPa calculated for photon energies up to 70 eV. The study of the optical functions may help to give a better understanding of the electronic structure. The imaginary and real parts of the dielectric function calculated are displayed in Fig.4. The calculated static dielectric functions ��1(0) are 8.585, 6.888, 5.652 and 4.264 for Na-hP4 phase at 260, 320, 400 and 600 GPa, respectively. The real part of the dielectric function ��1(��) reaches a peak value at 2.369, 3.090, 4.149 and 5.429 eV for Na-hP4 at different high pressures, and they are mainly generated by the electron transition from the top of the valence band to the bottom of conduction band. It is well known that the imaginary part of the dielectric function ��2 is the fundamental factor of the optical properties for any material. From the imaginary part of the dielectric function ��2 as a function of photon energy in Fig.4, we can see that the absorption starts at about 1.22, 1.76, 2.43 and 3.91 eV for Na-hP4 phase at 260, 320, 400 and 600 GPa, respectively, which is related to the minimum band gap at the �� point, corresponding to the transition ��V����C.
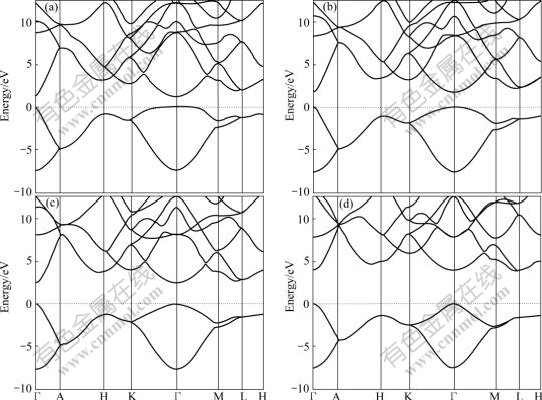
Fig.2 GGA-calculated band structure of Na-hP4 high pressure phase along some high-symmetry lines in Brillouin zone at different high pressures (Zero of energy is at Fermi level): (a) 260 GPa; (b) 320 GPa; (c) 400 GPa; (d) 600 GPa
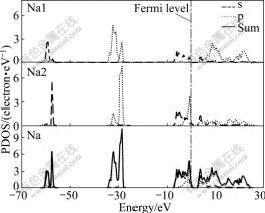
Fig.3 PDOS of Na1 2a (0, 0, 0), Na2 2d (2/3, 1/3, 1/4) and Na at 320 GPa
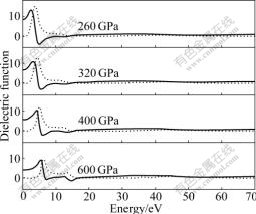
Fig.4 Imaginary part (dot line) and real part (solid line) of dielectric function of Na-hP4 at different high pressures
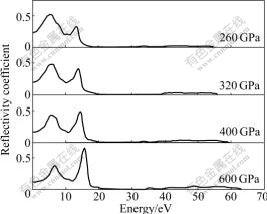
Fig.5 Reflectivity coefficients of Na-hP4 at different high pressures
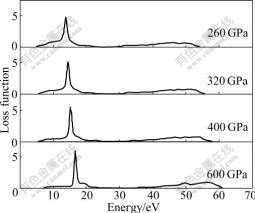
Fig.6 Loss functions of Na-hP4 at different high pressures
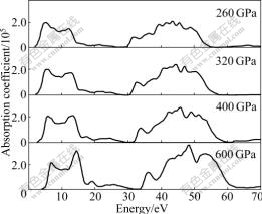
Fig.7 Absorption coefficient of Na-hP4 at different high pressures
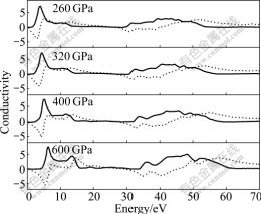
Fig.8 Imaginary part (dot line) and real part (solid line) of conductivity of Na-hP4 at different high pressures
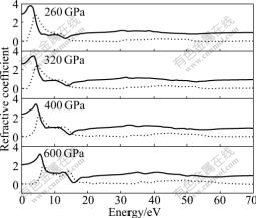
Fig.9 Imaginary part (dot line) and real part (solid line) of refractive coefficient of Na-hP4 at different high pressures
Fig.5 shows the reflectivity of Na-hP4 as a function of photon energy. We notice that there are two reflectivity peaks for Na-hP4 at different high pressures. And the first reflectivity peak decreases while the second peak increases with the pressure increasing, which is attributed to the change of the dielectric properties at different pressures. And the positions of the peaks also shift to the higher energy regions.
L(��) is an important factor describing the energy loss of a fast electron traversing in a material. The peaks in L(��) spectra represent the characteristic associated with the plasma resonance and the corresponding frequency is so-called plasma frequency. In addition, the peaks of L(��) also correspond to the trailing edges in the reflection spectra[17-18]. The peaks of L(��) for Na-hP4 phase presented in Fig.6 are at 13.734, 14.399, 15.111 and 16.556 eV, respectively, which correspond to the abrupt reduction of R(��) in Fig.5 at different high pressures.
The absorption coefficient is a parameter that tells the decay of light intensity spreading in unit distance in medium. The calculated absorption coefficients at different high pressures are presented in Fig.7. There are two absorption regions in the calculated photon energy range, which indicates special absorption characters of the Na-hP4 high pressure phase. From Fig.7, it can be seen that the absorption of light is almost zero when the energy is below 1.25, 1.77, 2.42 and 3.89 eV for Na-hP4 phase at 260, 320, 400 and 600 GPa, respectively. When the photon energy is more than these values, the absorption coefficient will increase, which corresponds to the calculated direct band gap of 1.258, 1.808, 2.473 and 3.932 eV of Na-hP4 phase at different high pressures, respectively. There are many peaks within the energy range studied, and the peak structures can be explained from the interband transitions using our band structure results stated above.
The imaginary part and real part of the optical conductivity are presented in Fig.8. The real part of the optical conductivity is zero until the photo energy increases to about 1.21, 1.75, 2.37 and 3.85 eV for Na-hP4 phase at 260, 320, 400 and 600 GPa, respectively. This is in accordance with the band gaps of the band structure, DOS and PDOS calculations.
According to the relation of refractive index and
dielectric function n2-k2=��1 and 2nk=��2, the refractive index n and extinction coefficient k of Na-hP4 are obtained. The calculated results are shown in Fig.9. The static refractive index n0 are 2.930, 2.624, 2.377 and 2.065 for Na-hP4 phase at 260, 320, 400 and 600 GPa, respectively, which agrees with the calculated result in Fig.4, where the corresponding calculated static dielectric function values ��1(0) are 8.585, 6.888, 5.652 and 4.264 ( from n02=��1(0)). The extinction coefficient k indicates the absorption of light, and at the same time, it also shows a great absorption characteristic at band-edge. The interband transitions discussed above also explain the origin of the peak structures in the refractive index and the extinction coefficient.
To the best of our knowledge, there are no experimental results on the optical properties of Na-hP4 high pressure phase. We therefore hope that our calculations will motivate experimental studies in this direction. Availability of experimental data will help in making quantitative comparisons with theoretical results.
4 Conclusions
1) A detailed investigation of the band structure, DOS, PDOS and optical properties of Na-hP4 phase at 260, 320, 400 and 600 GPa using the DFT-GGA method was carried out.
2) The band structure, DOS and PDOS of Na-hP4 phase were presented. The calculated band gap increases and the energy band expands to some extent with the pressure increasing.
3) The dielectric function, reflectivity, energy-loss function, absorption coefficients, optical conductivity, refractive index and extinction coefficient were calculated. The optical properties of Na-hP4 high pressure phase vary with the change of the pressure. Using the band structure and DOS (PDOS), the origin of the features that appear in the optical properties was discussed.
4) On the whole, our results for the optical properties of Na-hP4 phase await experimental findings for comparison.
References
[1] Neaton J B, Ashcroft N W. On the constitution of sodium at higher densities [J]. Phys Rev Lett, 2001, 86: 2830-2833.
[2] Hanfland M, Loa I, Syassen K. Sodium under pressure: bcc to fcc structural transition and pressure-volume relation to 100 GPa [J]. Phys Rev B, 2002, 65: 184109.
[3] McMahon M I, Gregoryanz E, Lundegaard L F, Loa I, Guillaume C, Nelmes R J, Kleepe A K, Amboage M, Wilhelm H, Jephcoat A P. Structure of sodium above 100 GPa by single-crystal X-ray diffraction [J]. Proc Natl Acad Sci, 2007, 104: 17297-17299.
[4] Christensen N E, Novikov D L. Calculated properties of high-pressure phases of simple metals [J]. J Phys: Condens Matter, 2002, 14: 10879-10883.
[5] Christensen N E, Novikov D L. High-pressure phases of the light alkali metals [J]. Solid State Commun, 2001, 119: 477-490.
[6] Gregoryanz E, Lundegaard L F, McMahon M I, Guillaume C, Nelmes R J, Mezouar M. Structural diversity of sodium [J]. Science, 2008, 320: 1054-1057.
[7] Ma Y M, Eremets M, Oganov A R, Xie Y, Trojan I, Medvedev S, Lyakhov A O, Valle M, Prakapenka v. Transparent dense sodium [J]. Nature, 2009, 458/460: 182-185.
[8] LI Yin-wei, MA Yan-ming. The novel behaviors of alkalis metals under high pressure [J]. Scientia Sinica Phys Mech & Astron, 2010, 40: 146-157.
[9] Payne M C, Teter M P, Allan D C. Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients [J]. Rev Mod Phys, 1992, 64: 1045-1050.
[10] Perdew J P, Zunger A. Self-interaction correction to density-functional approximations for many-electron systems [J]. Phys Rev B, 1981, 23: 5048-5079.
[11] Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method [J]. Phys Rev Lett, 1980, 45: 566-569.
[12] Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41: 7892-7895.
[13] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[14] Saha S, Sinha t p, Mookerjee A. Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3 [J]. Phys Rev B, 2000, 62: 8828-8834.
[15] CAI Meng-qiu, YIN Zhen, ZHANG Ming-sheng. First-principles study of optical properties of barium titanate [J]. Appl Phys Lett, 2003, 83: 2805-2807.
[16] SEGALL M D, LINDAN P J D, PROBERT M J, et al. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[17] SUN Jian, WANG Hui-tian, HE Ju-long, TIAN Yong-jun. Ab initio investigations of optical properties of the high-pressure phases of ZnO [J]. Phys Rev B, 2005, 71(12): 125132.
[18] Cheng Y C, Wu X L, Zhu J, Xu L L, Li S H, Chu Paul K. Optical properties of rocksalt and zinc blende AlN phases: First-principles calculations [J]. J Appl Phys, 2008, 103: 073707.
Foundation item: Project(50474051) supported by the National Natural Science Foundation of China
Corresponding author: SHI Yi-min; Tel: +86-15111157635; E-mail: hn_syb@126.com
DOI: 10.1016/S1003-6326(09)60262-4
(Edited by YUAN Sai-qian)
Abstract: The electronic structure and optical properties of novel Na-hP4 high pressure phase at different pressures (260, 320, 400 and 600 GPa) were investigated by the density functional theory (DFT) with the generalized gradient approximation (GGA) for the exchange and correlation energy. The band structure along the higher symmetry axes in the Brillouin zone, the density of states (DOS) and the partial density of states (PDOS) were presented. The band gap increases and the energy band expands to some extent with the pressure increasing. The dielectric function, reflectivity, energy-loss function, optical absorption coefficient, optical conductivity, refractive index and extinction coefficient were calculated for discussing the optical properties of Na-hP4 high pressure phase at different pressures.
