
��-����ϵ��������ѧ����
�й����������о�Ժ,�й����������о�Ժ �Ĵ�����621900 ,�Ĵ�����621900
ժ Ҫ��
���� ����ϵ��������ѧ���ʽ������ۺ�����������һ���������� (fcc) �ṹ��������� , ��ԭ���ܽ��ھ����� , ռ�ݰ������϶λ (O) , �γ�fcc���Ӿ��� , ����ʱ�������ȷ������� ;�������е���ɢ·��ΪO T OԾǨ , ���ڷ�ͬλ��ЧӦ ;���⻯�����ͬλ�ط������ӽϴ� , ���ܵ��¶ȡ���Ũ�ȵ�������Ӱ�� ;�� ����ϵ��p c�����߱��ֳ����õ�ƺ̨�� , Pd H��Pd D�������ٽ�� , ��δȷ��Pd T�����ٽ�� ;������ �ⷴӦ����ѧ�벻ͬ�ľ����й� ;�� ���ϵ������ϻ�ЧӦ
�ؼ��ʣ�
��ͼ����ţ� O614
�ո����ڣ�2002-01-21
Physical and Chemical Properties for Palladium-Hydrogen System
Abstract��
The physical and chemical properties for Pd H system were reviewed. The crystal structure of pure Pd is of face centered cubic ( fcc ) and it is a hydrogen absorbing metal. The hydrogen atom dissolves in lattice and occupies octahedral interstitial position, and then forms hydrogen sublattice (fcc ) . Upon hydrogen absorption the lattice undergoes an isotropic expansion while retaining its fcc structure. The defects in lattice are mainly extended type. The diffusion path of hydrogen is O T O jumping and there is reverse isotopic effect accompanying with quantum effect. The separation factor of palladium hydride is relatively larger than other common metal hydride and is affected by temperature and hydrogen concentration. The pressure composition isotherm in palladium hydrogen system exhibits good plateau. Pd H and Pd D systems have their own critical point respectively, but it has not been in agreement about the critical point of Pd T system. The dynamical property for single crystal Pd H reaction associates with the specific crystal face exposured to hydrogen. Tritium aging effect exists in Pd T system.
Keyword��
palladium; hydrogen isotope; metal hydride;
Received�� 2002-01-21
�Խ���������о����кܳ�����ʷ�� ����1866��Graham���ѷ��ִ����������ܽ��ڹ��ɽ����ٲ���1868���״ο������ٰ�Ĥ�Ծ��������� ����Pd����һЩ�Ͻ�, ��Pd-Ag, Pd-Al��, �������⻯���ͬλ��ЧӦ����, ��밹�������ͬλ��������ռ����Ҫ�ĵ�λ, ����������ͬλ�ط��롢 �����������յ���ѡ����, ����˽����⻯���������ѧ���ʶԻ����о�����Ӧ���о�������Ҫ���塣 ��-����ϵ�����н���-����ϵ��һ��������, ����������ص�������ѧ����, ��������Ըߡ� ���⻯�Լ��������Ӵ�����ʡ� �����ص���������-����ϵ�ľ���ṹ�� �����ɢ�� ����ܽ��ȶ���ѧ�� ͬλ��ЧӦ���ﻯ���ʵ��о���״��
1 Pd�⻯��Ľṹ�����״̬
1.1 Pd�⻯��Ľṹ
Pd�������������� (fcc) �ṹ, ÿ��������4��Pdԭ��, 4����������϶λ�Լ�8����������϶λ (T) �� �������a0Ϊ0.3890 nm (298 K) , ����ʱ�������ȷ�������, ��ʼ�ձ���fcc�ṹ�� ��ϡ���� (����) ����, 298 KʱPdHx��a0Ϊ0.3894 nm, ��Ӧ���⺬��H/Pd��0.015�� 뭻����Ц�����������a0��0.4025 nm, ��pH2=0.1 MPa, H/Pd��0.7ʱ, a0=0.4040 nm�� �ڵ�һ����뭻����еľ������ͽ�����
����������ϵ�д������������, �����ͼ�ȼ���-Һ����ͼ������
1.2 Pd-H���״̬
���ܼ���Ľ��������Pd-H��ϵ���൱�����Ľ��ͼ�� (1) �����Pdʱ, �����ĵ�ɴ�����Pdԭ�������䵽����Pd��Hԭ������ ����H��1s�����Pd��4d���֮�������ò������µĽ��״̬ (��s-d��ϵͼ���eV) �� Pdԭ����Χ�ĵ������d����, ��Hԭ����Χ�ĵ������s����, ����λ����Χ�ĵ�������൱; (2) Pd-H�����Ч�����Ƕ̾����, ��������������ڵ�Pdԭ��; (3) ��H���ӵ��ӻ��ı���4d���ӵ�״̬�ܶ�, ʹ֮��խ, �����ĵ���̬����, ��������������߽���ܷ����ƶ� (��������d �����) , ��������ʧ�˽����; (4) ����Fermi�ܼ��ĵ���̬�ܶȼ�С, Fermi������;�� (5) �ϸ���ܴ���������Ч��, ��Ϊֻ��һС�����ɶ���Hԭ���ṩ�ĵ���λ�ڵ���������
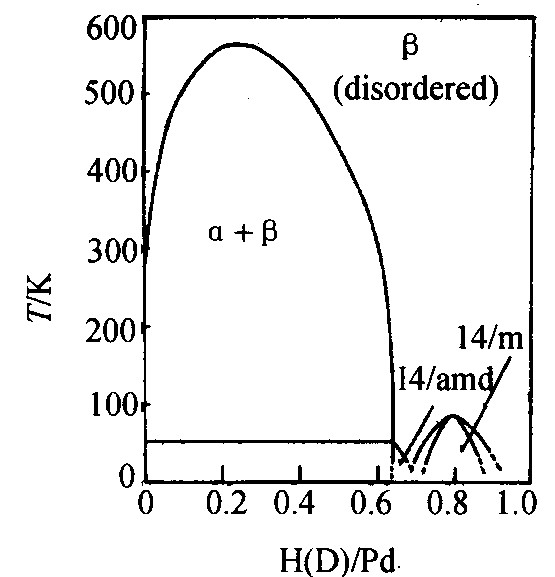
ͼ1 Pd-H (D) ��ϵ��ͼ
Fig.1 Phase diagram of Pd-H (D) system
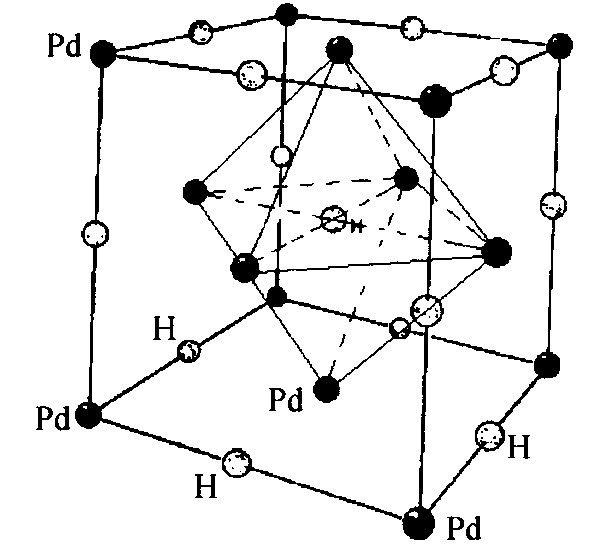
ͼ2 H (��D) ԭ���ڦ���Pd-H (��Pd-D) �е�λ��ģ��
Fig.2 Position modeling of H and D atoms in Pd lattice for ��-phase Pd-H (D)
1.3 ���ڦ������⻯���е�λ��
��-����ϵ������һֱ�������ǵ���Ȥ, ��Ҫ����Ϊ����ԭ���ڽ����з�ͬѰ������ɢ���ԡ� Davis
2 �������е��ܽ�
2.1 ���������ܽ�����۸���
�ڽ�������IJ�ͬ����������, �ܽ����һ��������Ҫ�����ʡ� ��ͬλ�� (H, D, T) �����е��ܽ���Ϊ�����Ƶ�, ������Ϊ���������ܽ⡣ ��������H, D, T��������Pd�����е�����ܺ����ܲ�ͬ, ���������е��ܽ�ȱ��ֳ����Ե�ͬλ��ЧӦ�� L
2.2 �������е��ܽ�ȵ�Ӱ������
2.2.1 Ӧ�������ܽ�ȵ�Ӱ��
���徲��ѧӦ������϶�ܽ�ȵ�Ӱ������Li��
�Ե�����Ӧ��, ���̻�ԭ�ɦ���H=1/3VH��x�� �˷���Ҳ����Ϊ���VH����������� VHΪH��Ũ�����ƫĦ�����
2.2.2 ȱ�ݵ�����
�ڳ���˻��Pd��, ���ϡ��������ܽ������Sieverts����, ��p1/2-rͼ�����Ե�, �ҵ�r����0ʱ����������ԭ���ཻ�������� ʵ�ʵ�ƫ���Ȼ������ȱ�ݵĴ��ڡ� �����ܽ�ȴ���Ӱ���ȱ���������ձ�����ڽ����е�ȱ��, �����ȱ�� (��λ������ԭ��) ����չȱ�� (λ��, ����, �����) ��
��������Pd�е��ܽ�������������䴦��ʹ��λ��Ӧ��������
Kirchheim
3 �����ٽ����е���ɢ
��������, ��ɢϵ������ƽ��λ�ü��ԾǨ��ʾ, ��:
D=fTa2/6��d (2)
ʽ��aΪƽ��ԾǨ����, ��dΪ����ԾǨ֮���ƽ��ʱ��, fT��У�����ӡ� ͼ3 (a) �������漰�������ֱ��ԾǨ��������ɢ�� Ea����ɢ���������Arrhenius��ʽ:
D=D0exp (-Ea/kBT) (3)
��ʵ��֤������˶�Ҫ���ӵöࡣ ͼ3 (b) ������һ�ֿ��ܵ�ԾǨ��ʽ, ��ʱ�۲쵽��Ea����������Dz�һ�µġ� ��������϶ԭ�� (��N, O, C) ���, Hԭ���ڽ����⻯���е��˶����൱���, �۲쵽��������ɢϵ��Dֵ��Χ�ڡ�10-16�͡�10-6 cm2��s-1֮�䡣
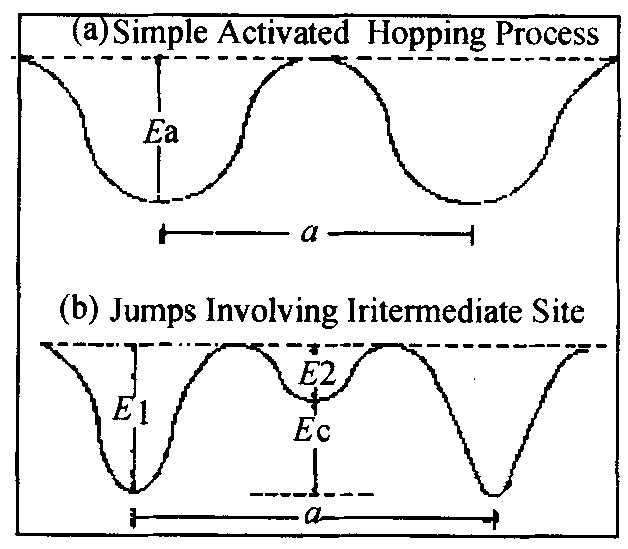
ͼ3 ��ɢ��������ͬλ�ص�����ʾ��ͼ
Fig.3 Sketch diagram of potential well of hydrogen isotopes during diffusion
3.1 ��ɢʵ����
��-����ϵ�е���ɢϵ���ѱ���ͬ�ķ���ȷ���� �������� �����ա� Gorsky������ ��������ɢ�� (QNS) �Լ���ϡ�������źϽ���˴ŷ�����
298 Kʱ, ����Pd�е���ɢϵ����Խϴ�, ����ĺ�����Сʱ, 뭺����Pd �е�Einstein��ɢϵ���ֱ�Ϊ:
D*H=2.5��10-7exp (-21.8 kJ/RT��xH) m2��s-1 (4)
D*D=1.7��10-7exp (-19.9 kJ/RT��xH) m2��s-1 (5)
298 Kʱ, H��D����ɢϵ���ֱ�Ϊ3.8��10-11��5.5��10-11 m2��s-1�� ���ܶ�H����ָǰ���Ӹ���, ��D����ɢϵ��ȴ��H��; H��D����ɢ���֮��IJ��������D��ɢ���졣 Bohmholdt��
������״̬�����Fick��ɢϵ��D (r) ֵ����ʵ�ʼ�ֵ��, ��298 Kʱ, �����е���ֵ����ϡ�����Լ4.5��
ʽ��r��H/Pdԭ�ӱ�, D*ΪEinstein��ɢϵ�� (��Ũ����) , aΪ��Ļ��, ��H����pH2�õ�:
3.2 ����������ɢ��ģ��ģ��
Gillan
Salomons
4 ��-����ϵ������ѧ����
4.1 Pd-Q��ϵ��ͼ
Pd-Q����͵����������ٽ�����³������ֹ��� (��ͼ1) , ��-����ϵ���ٽ�����1�� Pd-T�Ƿ�����ٽ���д�����, ��ԭ���������밵�Σ����������ʵ��ⶨ��ʵ�֡�
������һֱ��Ϊ��ͼ�Ļ��ܴ��������������ͼ, ������������ѧ����������Ҫ��Ĺ��� (��T��0ʱ, ������ʧ) ì�ܡ� 1978��Ross��
4.2 Pd-Q��Ӧ����ѧ��ʵ����
Pd-H��ϵ������ѧ�о������Ƿ����, ��Ϊ��Ļ�ѧλ������������ѹ��ȷ��, ����ѹ���뺬�������ƽ�⡣ ��ʵ��ȷ���ĵ����߿ɵõ���H, ����H=0.5RTlnpH2���¶ȵı仯, �ɵõ���ص�ƫĦ���ʺ�ƫĦ����, ����HH�ͦ�SH�� ���������ڵ���������, ���� (8) , (9) ����:
(?lnp1/2/? (1/T) ) r=��HH/R (8)
T (?lnp1/2/?T) r+lnp1/2=-��SH/R (9)
�������ദ��ƽ��̬, ƺ̨ѹ�����¶ȵı仯���Ƶ�����Ӧ (10) ��ƺ̨����ѧ��:
ʽ��a��b�ֱ�Ϊϡ������⻯�������ɡ� ���������p-c-T��������Լ�L
��1 ��-����ϵ���ٽ糣��
Table 1Critical parameter of Palladium-Hydrogen system
| ��ϵ | �ٽ糣�� | ||
| �ٽ��¶� Tc/K |
�ٽ�ѹ�� pc/MPa |
�ٽ�Q/Paԭ�ӱ� rc (Q/Pd) |
|
| Pd-H | 563 | 1.9 | 0.257 |
| Pd-D | 556 | 3.9 | 0.25 |
| Pd-T | ���ٽ糣��, �����ڷ��� | ||
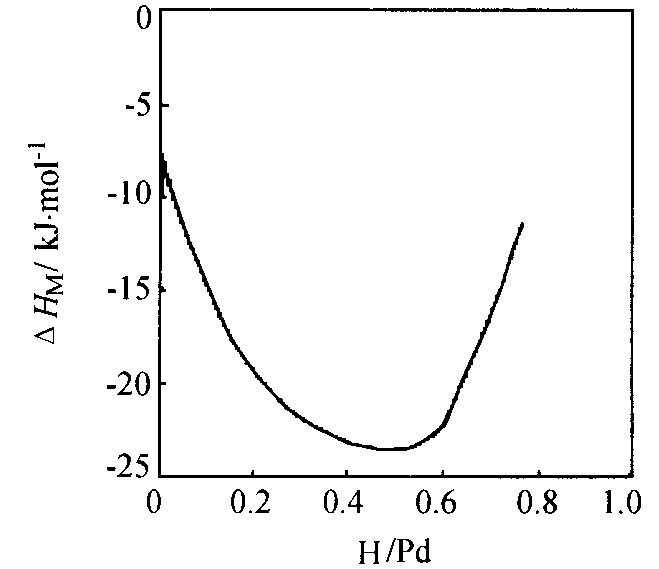
ͼ4��5�����˦�HH�ͦ�SH��r�ı仯, ���Կ�����HH�ͦ�SH��������Сֵ�� ��ʱϣ�����¶�Ӧ����Tc (����ѧ���ſ�����r����������) , ����ͼ4��5, �¶ȱ���Ϊ��ѡ����500 K, ���Լ����⻯���δ�γɡ�
����ϡ��ʱ���ܽ�����ƫĦ���ʦ�H
��2 Pd-H��Pd-D����ƺ̨�����ʺ���
Table 2Enthalpy and entropy of plateau phase for Pd-H and Pd-D
| Hf/ (kJ��mol-1) | Hd/ (kJ��mol-1) | Sf/ (kJ��mol-1) | Sd/ (kJ��mol-1) |
| ���ػ����� | |||
| -18.70.15 (H) | 19.50.25 (H) | -46.30.4 (H) | 46.20.6 (H) |
| -16.80.30 (D) | 17.70.25 (D) | -46.70.8 (D) | 46.70.65 (D) |
| ������ | |||
| -19.090.15 (H) | 19.280.20 (H) | -46.6 (H) | 46.6 (H) |
| -17.270.25 (D) | 17.230.25 (D) | -46.6 (D) | 46.4 (D) |
ͼ4 HH��r�ı仯
Fig.4 Variation of HH with H/Pd=r
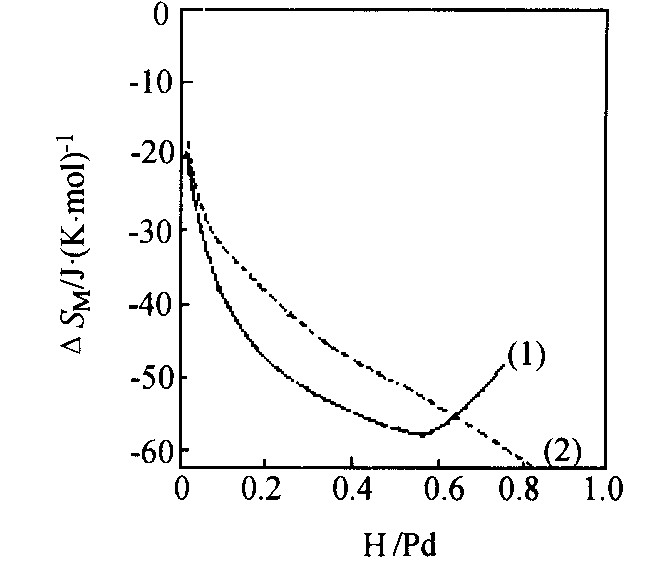
ͼ5 ��SH��r�ı仯
Fig.5 Variation of ��SH with H/Pd=r (1) ʵ������; (2) ����״̬
ֱ�Ӵӷ�Ӧ����ȷ���ʱ�����p-c-T������ ͼ7��ʾ��298 Kʱ������ƽ̨������һ�������ɷ�Ӧ���������������� (뭡� �) ������ʱ�
�����ȼ����ƺ̨�����ʼ������¶��ء� ����, Picard��
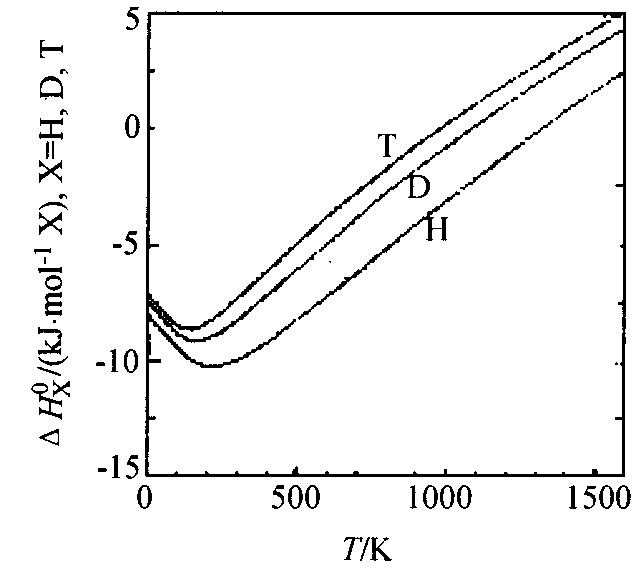
ͼ6 ��H0�����¶ȵĺ�����ϵͼ
Fig.6 Plot of ��H
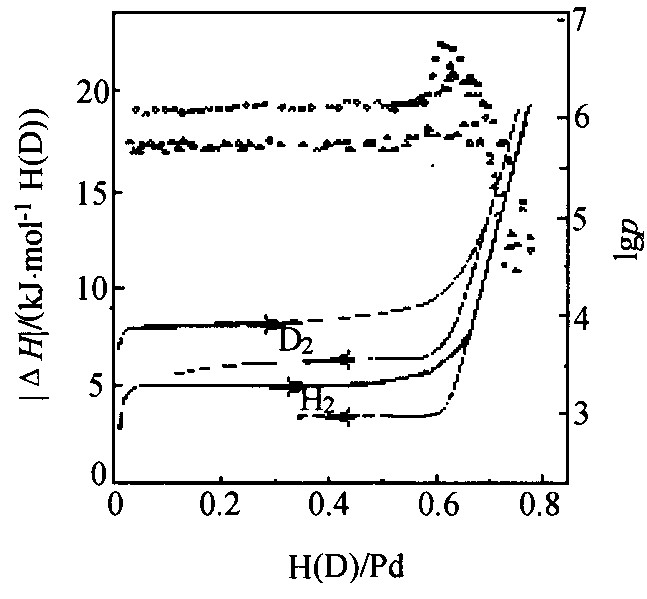
ͼ7 ���ȷ�ȷ����298 KʱPd-H��Pd-D��ϵ���ʱ�
Fig.7 Relative enthalpies for both Pd-H and Pd-D systems determined from reaction calorimetry (298 K) using Pd foil
ʽ�е�|��Hplat|������ֵ,
5 Pd�⻯���ͬλ��ЧӦ
5.1 Pd�⻯������ͬλ��ЧӦ��һ�����
L
ͬλ�ط������� (����H��D) ������Ϊ:
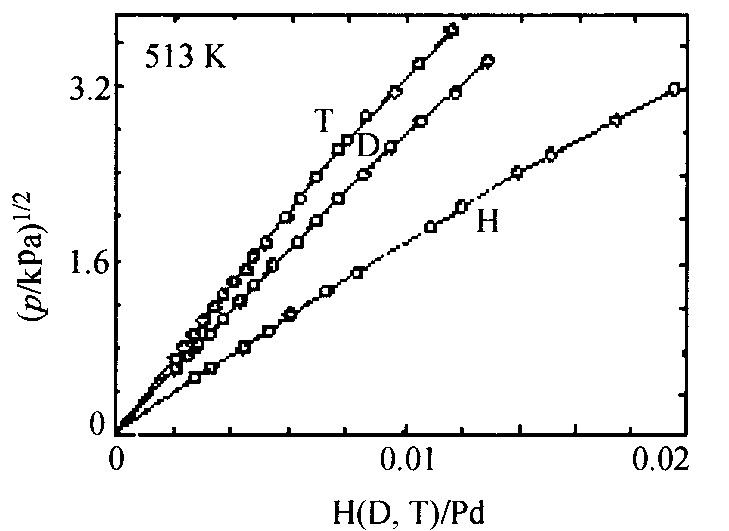
ͼ8 �������ܽ�ȵ�ͬλ��ЧӦ
Fig.8 Isotopic effect of solubility in ��-phase for palladium-hydrogen systems
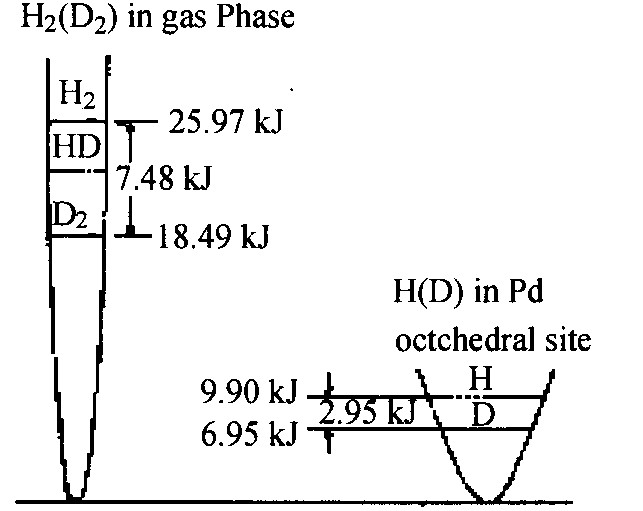
ͼ9 �����������H, D����ܼ������λ��
Fig.9 Relative location of the zero point energy levels of H and D in gas phase and dissolved in ��-phase of palladium
ʽ��x�������� (g) ����� (s) �еĸ�����ͬλ�ص�Ħ�������� ������ͬλ�ط������ӱ���ʽδ�漰��-���������ͬλ���������ʵĹ�ϵ�� ����Brodousky��
5.2 Pd�⻯�����ͬλ�ط�������
Pd�����൱���ͬλ��ЧӦ (��������) , �Դ˽��������ױ����� ��Pd-H��ϵ�ķ������ӽ����������IJ�������Fukada��
����ƽ�ⳣ���ͷ������ӵĹ�ϵ:
��=K/R (13)
��H-D������ӦK=0.5�� ƽ�ⳣ���뷴Ӧ�ı�������G0�Ĺ�ϵ����ʽ����,
��G0=-RTlnK (14)
���ǿɵõ�ln��= (��H/T-��S) /R�� ����ģ�Ͷ���������Ч��, ��Ϊ�ɴӾ�������ѧ�Ƶ������� ����, ��Pd-H��ͼ�Ħº�������, �ⲻ���Դ���Ԫ��ʽ����, ���Ƿֱ��Ի����P����Ԫ�ͻ����ﹲ�����ʽ����, ����������Һ���������Dz��ϸ��, ����ķ����������¶ȵĹ�ϵ����ͬ��ǰ�����ʽ�� ��ͬ���߸����˷������� (��) ���¶ȵĹ�ϵ���ڱ�3�� ��Ƚ϶���, ����ͬ�¶���, H-T�����ķ������Ӹ���, D-T�ķ�������С��H-D�� H-T��D-T�ķ�������ʾ��ͼ10
��3 �������� (��) ���¶� (T) �Ĺ�ϵ
Table 3Variation relation of separation factor �� with temperature
| ��-T�Ĺ�ϵ����ʽ | �ο����� |
| ��H2-HD=exp (228/T-0.121) | 32 |
| lg��=225/T-0.51 | 33 |
| lg��=215/T-0.47 | 34 |
| ln��=245/T+0.055 | 32 |
| lg��=119/T-0.07 (����D��ɽӽ�0%) lg��=82/T+0.025 (����D���Ϊ70%) |
35 |
| ��=exp[-0.3+B (xH) (��1000T) 0.75] B (xH) =0.50916-0.0037952xH-0.15589x2�� |
36 |
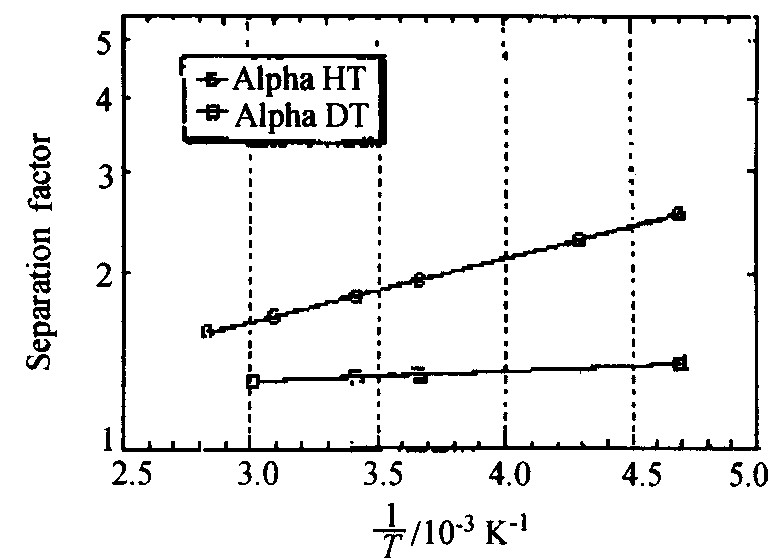
ͼ10 H-T��D-T�ķ����������¶ȹ�ϵ
Fig.10 Relationship between separation factor of H-T and D-T and temperature
6 Pd-H��Ӧ����ѧ�����
�о����뵥��Pd�IJ�ͬ���������õķ�����: ���Ѹ��� (TDS) �� ���ܵ������� (LEED) �� ����ɢ�估���ۼ��㡣 �۲쵽�ķ�Ӧ��Ϊ�뱩¶���������ض������йء� ����120 Kʱ, (110) �����������γ������� (2��1) �ṹ, ����һ������, �����Ƕ�����ʱ, (1��2) ���ع����濪ʼ����
7 ��-���ϵ����ϻ�ЧӦ
����밹��յĽ����⻯����ھ����л����˥�����3He�� ��3He����Ľ����⻯�ᄃ��ṹ�仯���½����⻯������������ܱ仯�� Nobile��
Pd������������۲쵽��ѹ���仯���¶ȵĺ���, ��������3He�ۻ����¾���ṹ�仯����ġ� ��ǰ��3He�ͷŵIJ���
8 ������
��������, ����ѧ�߶���-����ϵ���о��ǽ�Ϊ�����, ����һЩ��������ķ�����Ȼ����, ���ݵIJ���Ҳ�Ƚ�����, �ٵ����ȼ����ȶ���-����ϵ���ȶ���ѧ�� �����ɢ���ԡ� ��ͬλ�ط��������Լ��ܽ�ȵ��ﻯ���ܶ�����Ӱ��, ��ͬλ���ڲ�ͬ��̬�ٱ���Ľ����������д���һ���о�, �ٵ���ϻ�ЧӦ���ۻ����Բ���ȫ���, ���, ����-����ϵ��ϵͳ�о�����������롣
�����
[1] ��BaabaI , HardyP , SanMartinA , etal.��J .Phys., 1987, 17:2041.
[2] ��ShirberJE , MorosinB .��Phys.Rev., 1975, B12:117.
[3] ��AbbensethR , WipfH .��J .Phys., 1980, F10:353.
[4] ��AlefeldG .��Ber.BunsengesPhys.Chem., 1972, 76:746.
[5] ��AdersonIS , RossDK , CarlileC .��Phys.Lett., 1978, 68A :249.
[6] ��BlaschkoO , FratzlP , KlemencicR .��Phys.Rev., 1981, B81:277.
[7] ��BondR , RossDK .��J .Phys.1982, F12:597.
[8] ��RushJ , RoweJ , RichterD .��Z .Phys., 1984, B55:283.
[9] ��RoweJ , RushJ , SmithH , eta.��Phys.Rev.Lett., 1974, 33:1297.
[10] ��DavisWD .��KnollsAtomicPowerLab.Rep., 1954.1227.
[11] ��WorshamJE , WilkisonMK , ShullCG .��J .Phys.Chem.SolidsPergamonPress, 1957, 3:303.
[12] ��L��asserR , PowellGL .��Phys.Rev., 1986, B34 (2) :578.
[13] ��LiJ , OrianiR , DarkenL .��Z .Phys.Chem., 1966, 49:271.
[14] ��WriedtH , OrianiR .��ActaMetall., 1970, 18:271.
[15] ��KirchheimR .ActaMetall., 1986, 34:37.
[16] ��ManzkeR , CreceliusG , FinkJ , etal.��J .Phys., 1982, F12:L279.
[17] ��GuthrieS , WolferW .��J .LessCommanMet., 1990.
[18] ��BesenbacherF , MyersS , NorskovJ.��Nucl.Instr.Meth.Phys.Res., 1985, B78:55.
[19] ��KirchheimR .��ActaMetall.1982, 30:1069.
[20] ��LynchJ, ClewleyJD , CurranT , etal.��J .LessCommanMet., 1977, 55:153.
[22] ��KirchheimR .��Prog.Mater.Sci., 1988, 32:261.
[23] ��BohmholdtG , WickeE .��Z .Phys.Chem., 1967, NF56:133.
[24] ��GillanMJ .��J .Phys., 1990, C 19:6169.
[25] ��CulvahouseJ , RichardsP .��Phys.Rev., 1988, B 38:10020.
[26] ��SalomonsE .��J .Phys.Condens.Matter., 1990, 2:845.
[27] ��L��asserR , PowellGL .��Phys.Rev., 1986, B 34:578.
[28] ��FlanaganT , LuoW , ClewleyJ .��J .LessCommonMet., 1991.
[29] ��PicardC , KleppaOJ , BoureauG .��J .Chem.Phys., 1978, 69:5549.
[30] ��L��asserR .��Phys.Rev., 1984, B 29:748.
[31] ��BrodowskyH , RepenningD .��ZPhys.Chem., 1979, NF114:141.
[32] ��FukadaS , FuchinoueK , NishikawaM .��JournalofNuclearMa terials, 1995, 226:311.
[33] ��ThomasCO , SmithHA .��JournalofPhsicalChemistry, 1958, 63:427.
[35] ��BotterF , MenesJ , TistchenkoS , etal.��BulletindelaSoci����ChimiquedeFrance, 1965, 11:3374.
[36] ��ThomasManuelOrtiz.��LA -13454-T , DE98006025, LosAlamosNationalLaboratory.1998.
[37] ��AndreevBM , PerevezentsevAN , MandrykinIA , etal.Radio khimiya, 1986, 28 (2) :212.
[38] ��CattaniaM , PenkaV , BehmR , etal.��Surf.Sci., 1983, 126:382.
[39] ��HeJ , HarringtonD , DriffithsK , etal.��Surf.Sci., 1988, 198:413.
[40] ��FelterT , SowaE , VanHoveM .��Phys.Rev., 1989, B40:891.
[41] ��KayB , PedenC , GoodmanDW .��Phys.Rev., 1986, B 34:817.
[42] ��AuerW , GrabkeH .��Ber, BunsengesPhys.Chem., 1974, 78:58.
[45] ��ThomasGJ , SwansigerWA , BaskesMI.��J .Appl.Phys., 1979, 50:6942.
[46] ��CoronadoPR , FearonEM , GarzaRG , etal.��FusionTechn ol, 1988, 14:741.
[47] ��ThomasGJ , MintzJM .��J .Nucl.Mater., 1983, 116:336.
[48] ��LasserR , Bickman, K , Trinkaus.��Phys.Rev., 1989, B 40:3306.
[49] ��WilsonWD , BissonCL , BaskesML .��Phys.Rev., 1981, B24:5616.
[1] ��BaabaI , HardyP , SanMartinA , etal.��J .Phys., 1987, 17:2041.
[2] ��ShirberJE , MorosinB .��Phys.Rev., 1975, B12:117.
[3] ��AbbensethR , WipfH .��J .Phys., 1980, F10:353.
[4] ��AlefeldG .��Ber.BunsengesPhys.Chem., 1972, 76:746.
[5] ��AdersonIS , RossDK , CarlileC .��Phys.Lett., 1978, 68A :249.
[6] ��BlaschkoO , FratzlP , KlemencicR .��Phys.Rev., 1981, B81:277.
[7] ��BondR , RossDK .��J .Phys.1982, F12:597.
[8] ��RushJ , RoweJ , RichterD .��Z .Phys., 1984, B55:283.
[9] ��RoweJ , RushJ , SmithH , eta.��Phys.Rev.Lett., 1974, 33:1297.
[10] ��DavisWD .��KnollsAtomicPowerLab.Rep., 1954.1227.
[11] ��WorshamJE , WilkisonMK , ShullCG .��J .Phys.Chem.SolidsPergamonPress, 1957, 3:303.
[12] ��L��asserR , PowellGL .��Phys.Rev., 1986, B34 (2) :578.
[13] ��LiJ , OrianiR , DarkenL .��Z .Phys.Chem., 1966, 49:271.
[14] ��WriedtH , OrianiR .��ActaMetall., 1970, 18:271.
[15] ��KirchheimR .ActaMetall., 1986, 34:37.
[16] ��ManzkeR , CreceliusG , FinkJ , etal.��J .Phys., 1982, F12:L279.
[17] ��GuthrieS , WolferW .��J .LessCommanMet., 1990.
[18] ��BesenbacherF , MyersS , NorskovJ.��Nucl.Instr.Meth.Phys.Res., 1985, B78:55.
[19] ��KirchheimR .��ActaMetall.1982, 30:1069.
[20] ��LynchJ, ClewleyJD , CurranT , etal.��J .LessCommanMet., 1977, 55:153.
[22] ��KirchheimR .��Prog.Mater.Sci., 1988, 32:261.
[23] ��BohmholdtG , WickeE .��Z .Phys.Chem., 1967, NF56:133.
[24] ��GillanMJ .��J .Phys., 1990, C 19:6169.
[25] ��CulvahouseJ , RichardsP .��Phys.Rev., 1988, B 38:10020.
[26] ��SalomonsE .��J .Phys.Condens.Matter., 1990, 2:845.
[27] ��L��asserR , PowellGL .��Phys.Rev., 1986, B 34:578.
[28] ��FlanaganT , LuoW , ClewleyJ .��J .LessCommonMet., 1991.
[29] ��PicardC , KleppaOJ , BoureauG .��J .Chem.Phys., 1978, 69:5549.
[30] ��L��asserR .��Phys.Rev., 1984, B 29:748.
[31] ��BrodowskyH , RepenningD .��ZPhys.Chem., 1979, NF114:141.
[32] ��FukadaS , FuchinoueK , NishikawaM .��JournalofNuclearMa terials, 1995, 226:311.
[33] ��ThomasCO , SmithHA .��JournalofPhsicalChemistry, 1958, 63:427.
[35] ��BotterF , MenesJ , TistchenkoS , etal.��BulletindelaSoci����ChimiquedeFrance, 1965, 11:3374.
[36] ��ThomasManuelOrtiz.��LA -13454-T , DE98006025, LosAlamosNationalLaboratory.1998.
[37] ��AndreevBM , PerevezentsevAN , MandrykinIA , etal.Radio khimiya, 1986, 28 (2) :212.
[38] ��CattaniaM , PenkaV , BehmR , etal.��Surf.Sci., 1983, 126:382.
[39] ��HeJ , HarringtonD , DriffithsK , etal.��Surf.Sci., 1988, 198:413.
[40] ��FelterT , SowaE , VanHoveM .��Phys.Rev., 1989, B40:891.
[41] ��KayB , PedenC , GoodmanDW .��Phys.Rev., 1986, B 34:817.
[42] ��AuerW , GrabkeH .��Ber, BunsengesPhys.Chem., 1974, 78:58.
[45] ��ThomasGJ , SwansigerWA , BaskesMI.��J .Appl.Phys., 1979, 50:6942.
[46] ��CoronadoPR , FearonEM , GarzaRG , etal.��FusionTechn ol, 1988, 14:741.
[47] ��ThomasGJ , MintzJM .��J .Nucl.Mater., 1983, 116:336.
[48] ��LasserR , Bickman, K , Trinkaus.��Phys.Rev., 1989, B 40:3306.
[49] ��WilsonWD , BissonCL , BaskesML .��Phys.Rev., 1981, B24:5616.
