
�й���ɫ����ѧ�� 2004,(11),1969-1976 DOI:10.19476/j.ysxb.1004.0609.2004.11.030
������ͪ��(��)�Ļ�ѧ����
����������о������ҹ�������Ϲ��̼����о�����,����������о������ҹ�������Ϲ��̼����о�����,����������о������ҹ�������Ϲ��̼����о�����,����������о������ҹ�������Ϲ��̼����о�����,����������о������ҹ�������Ϲ��̼����о�����,���������Ƽ�����˾ ����650221 ,����650221 ,����650221 ,����650221 ,����650221 ,����650106
ժ Ҫ��
�ϳ���������ͪ��,���������⡢���⡢�˴ź������ֶ��о���������ͪ��(��)�Ļ�ѧ�ṹ�������������:������ͪ�����ڵ������Ķ��������;���������������м����γ�,�����ʻ��Ѳ��߱����͵��ʻ�����;�ڿ�ԭ�ӵĺ����,������ͪ�����λ����һ����,�ͷų������������ͪ,����γ�103Rh+,ͬʱ���湲�ۼ��Ķ���������;�ڴų���,����Rh3+��ǿ������ЧӦ�Լ����ϻ��Ļ�������ЧӦ,H�˻�ѧλ����ͳ��ƶ���ͨ��TG-DTA��GC-MS������̽����������ͪ����ȷֽ���Ϊ,Ϊ��Ӧ���ṩ����ѧ���ݺ����۲ο�,��������300����Ϊ��������ͪ��Ϊǰ�����CVD��������յij����¶ȡ�
�ؼ��ʣ�
������ͪ��;�ṹ����;�ȷֽ�;��ѧ�������;
��ͼ����ţ� O627.8
����飺����(1981),��,˶ʿ�о���;��ΰƽ,�о�Ա;�绰:0871 5133648;E mail:cuiliang1234@163.com;
�ո����ڣ�2003-12-12
��������Ժ����������������Ŀ(2004EG115047);����ʡԺ����������������Ŀ(2003KFZX 15);
Chemical properties of rhodium(��) acetylacetonate
Abstract��
Rhodium(��) acetylacetonate ��Rh(acac)3��, a precursor for preparing Rh layers by CVD technique, was prepared. Its spectroscopic properties were investigated via UV-vis, IR, ()1HNMR and MS and its thermal decomposition behavior via TG-DTA and GC-MS. The results show that, Rh(acac)3 is a low spinning and inert coordination compound, the characteristics of carbonyls in the ligand disappear due to the formation of conjugate �� bonds in Rh3+��acac- chelating ring, after bombarded by fast atoms, the coordination bonds break and the organic covalent bonds are splited with the formation of ()103Rh+, chemical shifts of H nuclei go downward to low field because of electron-attraction of Rh3+ and electron-cyclic effect in the chelating rings, and it sublimates and decomposes at (247 ��) and 245 �� in air and argon, respectively. Series of new complexes, different with the temperatures, appear in the course of the decomposition, according to which it is suggested that a better deposit temperature should be (300 ��) when plating Rh layers via CVD technique with Rh(acac)3 as the precursor.
Keyword��
rhodium(��) acetylacetonate; chemical structure; thermal properties; CVD;
Received�� 2003-12-12
��Cl-�� SO
������ͪ��(��)[Rh(acac)3]��һ�����͵Ĺ�����л���λ����� ���Ż�ѧ�����������(CVD)����������, ������ͪ�������ӷ��� �ֽ⡢ �����ڶ����л��ܼ����ص�
Rh(acac)3�ĺϳɷ����������������б���
1 ʵ��
1.1 �ϳ�
ȡˮ�����Ȼ���(���������38%, ���Ϲ��в�ҵ�ɷ�����˾�ṩ)1.00 g, ��50 mLˮ�ܽ�, ��4 mol/L��NaOH(������)��Һ10 mL������Rh(OH)3, ���ˡ� ϴ�Ӻ���ϡ�����ܽ�, �õ���������Һ, Ȼ��������Hacac(�Ϻ���ѧ�Լ���, ������)�������, ������ɫ��Rh(acac)3����, �����ռ�����ˮ�� �Ҵ�ϴ��, ��70 ���º��, �õ�1.16 g�IJ�Ʒ, ����Ϊ76%�� ����Rh(acac)3��Ʒ��Ԫ�ط������ΪC 45.00%�� H 5.29%�� Rh 25.44%, ��Rh(acac)3Ԫ�����ۺ���(C 45.01%�� H 5.25%�� Rh 25.72%)һ��(C�� HԪ�ط������й���ѧԺ�Ϻ�ҩ����������IJⶨ, Rh�������������в�ҵ��˾�������IJⶨ)��
1.2 �ṹ���Բ��Լ������ʲ���
��������ͪ��(��)��Ʒ�����ȷ���, �����ȷ�Ϊ�α�, �ڵ���UV-2201������ɼ��ֹ��ȼ��ϲⶨ����200��600 nm��Χ�ڵĵ��ӹ���ͼ, ������ȷ������max����Ӧ��; ���ȷ�Ϊ�ܼ�, ��DRX500�ͺ˴Ź�����(��ʿ)��¼��1HNMR��; �Ը���Ϊ����, ����FAB+��Autospec 3000�߷ֱ�������(Ӣ��)�ϲⶨ��MS; �������ײ���KBrѹƬ����FTS-135�ͺ��������(����)�ϲⶨ��
����-���ȷ�����������Ϊ�ձ���ѧ�����ʽ����������Thermoflexϵ���ȷ���װ�õ�TG-DTA���֡� ������ͪ���ڿ���������������������½���, �������ʶ�Ϊ10 K/min, ���ÿ�������������ʢװ��Ʒ, �α���Ʒ��Ϊ��-Al2O3��ĩ, �������ٶ�Ϊ50 mL/min��
Ϊ��һ���о�Rh(acac)3���ȷֽ���Ϊ, �ֱ��ڶ��ԺͿ������������½�����-�����÷���, �����ڲ�ͬ�¶���������Ʒ����Ҫ�ֽ��� �˷�����������ΪCDS PYROPROBE 2000�����ѽ��Ǽ�HP6890/MS5972����-�����÷�����(����)�� Rh(acac)3��Ʒ�趨��5���¶�Ϊ250�� 300�� 400�� 500�� 600 ��, �������µ�ָ���¶Ⱥ�10 s, Ȼ���ѽ��������������(���������)��������ɫ���з���, �ٽ��������м�⡣
2 ���������
2.1 ���ӹ���
ͼ1��ʾΪRh(acac)3���ȷ���Һ(5.80��10-5 mol/L)�е�����-�ɼ����չ��ס� ��ͼ��֪, Rh(acac)3������-�ɼ���������������ǿ���շ���һ�����, �����ɲ�-�ȶ���������ȼ��㹫ʽA=��bc (����AΪ�����; ��ΪĦ������ϵ��; cΪ��ҺĦ��Ũ��; bΪ��̳���, ��ʵ����b ֵΪ1 cm), �ɼ���������շ��Ħ������ϵ����max��

ͼ1 ������ͪ�����ȷ���Һ�е�����-�ɼ����չ���
Fig.1 UV-vis absorpbance spectra for chloroform solution of Rh(acac)3
������maxΪ320.3 nm(��maxΪ10 602.9 L��mol-1��cm-1)��240.6 nm(��maxΪ8 758.6 L��mol-1��cm-1)�����շ�, ��λ�ú�����ǿ������, Ӧ�������������ӵ�d��dԾǨ�� ������������[Na(acac)]�ĦС���*ԾǨ�������շ�����max=261 nm��
����Rh3+����d6���ӹ���
2.2 �������
������ͪ����������ͪ��ĺ��������1��
������ͪ���ֳ�2,5-���ͪ��, �������´������¹���ṹ, �����ڦе����Ƶ�����, �乲��ʽ��ϩ��ʽ�ı����������

���, ��IR������, �����������͵�CO���Գ����������շ�, ͪʽCO��ϩ��ʽCO, ǰ��Ϊ1 668.98 cm-1(m), ����������ϵ������, Ϊ1 623.79 cm-1(s)��
������ͪ����Rh3+��λ��,�γ��˴�м�, ���ϵ��������������ӵķ����ƶ�, ʹ��CO������, �Ѳ��߱����͵��ʻ�����, �����������շ岨���½���Լ100 cm-1�� ͬʱ, ���������м����γ�, ʹ������ԭ����ʽ�����ֲ�ͬ��CO��һ��, �ں�������ϱ���Ϊ1 569.39 cm-1��ǿ���շ塣 Acac-��λ���������M-O���൱��Ҫ��, ��Ϊ����ֱ���ṩ���й�M��O��ǿ�ȵ���Ϣ�� Nakamoto��
������������ͬλ��λ���о���������450 cm-1�������״���M��O��C��CH3������ģʽż�ϵ�, ���ڵ�Ƶ�����DZȽϴ����M��O�������״��� ����ʵ������(��������Ƶ�����ơ�ͬλ���о���), ���о�ֻ�ܴ������Ͻ���1�е�465.96 cm-1�����ΪRh��O����������C��CH3������ż��, ��434.84 cm-1�����ΪRh-acac���ı�����
��1 ������ͪ����������ͪ��ĺ������
Table 1 Infrared spectrum of Na(acac) and Rh(acac)3 (cm-1)
| Na(acac) | Rh(acac)3 | Corresponding | Na(acac) | Rh(acac)3 | Corresponding |
3 073.89 |
3 077.15 | ��(CH) | 1 012.09 |
1 022.71 | ��(CH3) |
| ��(CH3) | 912.28 |
936.70 | ��(CC)+��(CO) | ||
767.63 |
��(CH) | ||||
1 668.98 |
��(CO) | ��(C��CH3)+Ring deformation +��(Rh��O) |
|||
1 623.79 |
1 569.38 | ��(CO) | |||
1 510.09 |
1 518.64 | ��(CC) | 656.10 |
662.87 | |
1 451.81 |
��(C��H)+��(CC) | ||||
1 409.85 |
1 383.36 | ��(CH3) | 647.50 | Ring deformation+��(Rh��O) | |
1 235.37 |
1 270.94 | ��(C��CH3)+��(CC) | 465.96 | ��(Rh��O)+��(C��CH3) | |
1 205.09 |
1 201.82 | ��(C��H)+��(C��CH3) | 434.84 | Ring deformation |
2.3 ����
�ڿ�ԭ�Ӻ��(FAB+)��, ������ͪ������˷������ӷ�M+, ���ǿ��100%, �����ŷ��ѳ��ʺɱ�(m/e)Ϊ301(��Է��84%)�� 203(��Է��10%)��259(��Է��5%)����Ƭ��, ���ѽⷴӦ��ͼ2��ʾ��
�ɼ�, �ڿ�ԭ�Ӻ����, ��ǿ��Խ�������λ���ȶ���, ���ͷų�����, ����γ�103Rh+, ͬʱҲ�������л����ۼ��Ķ��ѡ�
ͬʱ��MS�ϻ�������m/eΪ493(��Է��3%)�����ӷ�, ���Խ���Ϊ������ͪ��ķ�������ʸ��ͷ���(C3H8O3)�Ӻ϶��ɡ� �ڿ�ԭ�ӵĺ����, Rh3+��3��������ͪ�е�����1����Rh��C3����λ
2.4 �˴Ź�������
Rh(acac)3��Na(acac)����ͼ
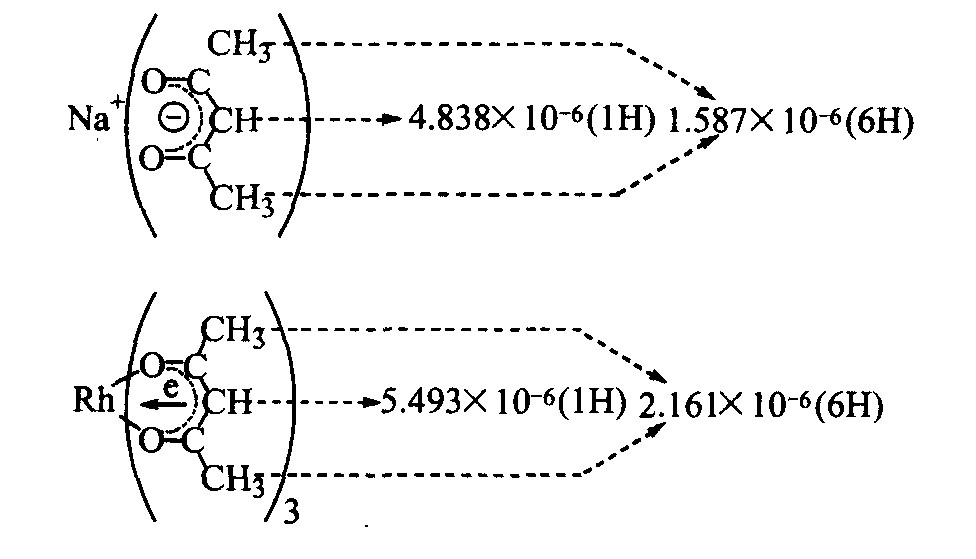
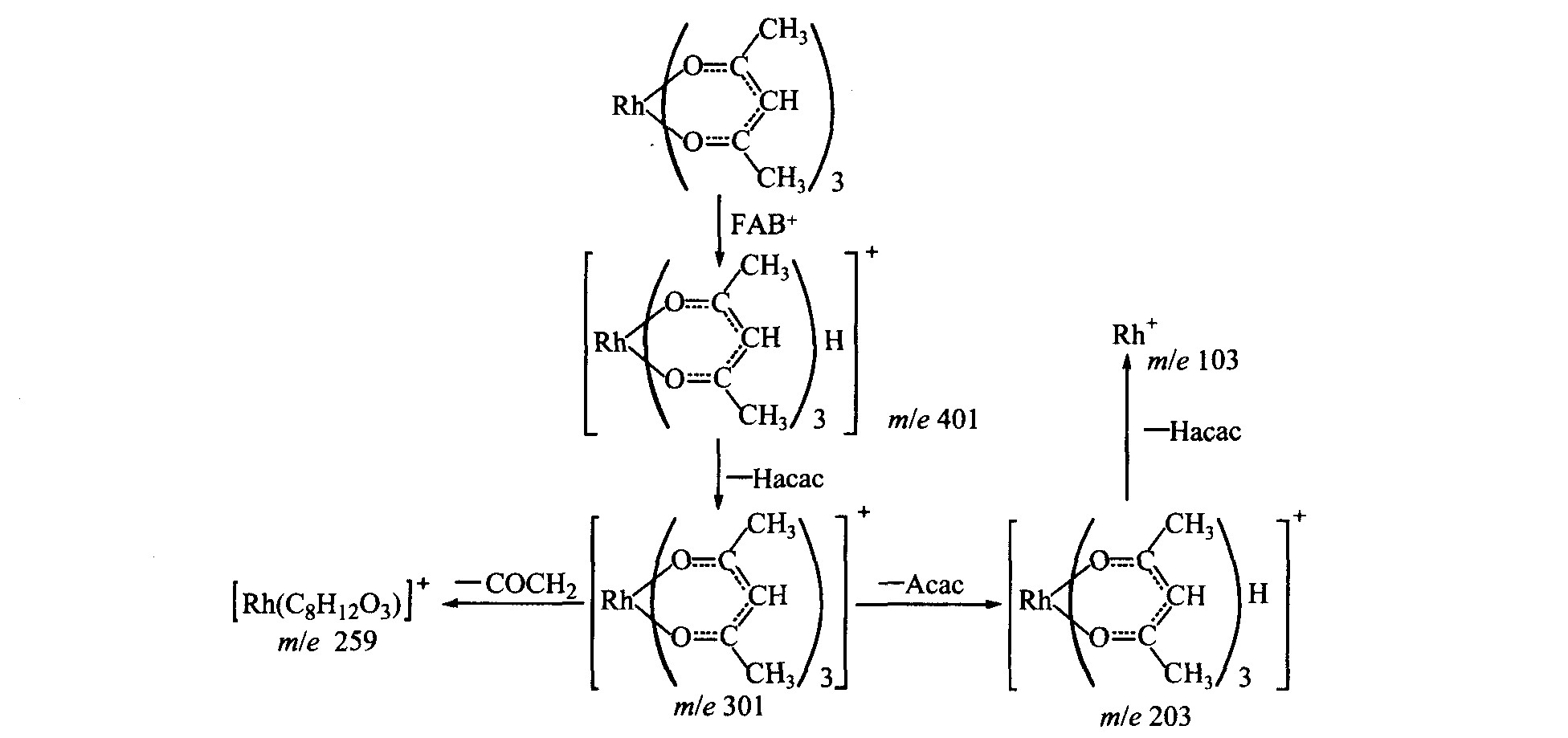
ͼ2 ������ͪ����������Ҫ��Ƭ���ӷ���γ�
Fig.2 Formation of main protonized peaks in MS of Rh(acac)3

ͼ3 ������ͪ�����Rh��C����λʾ��ͼ
Fig.3 Formation of Rh��C bonded chelate
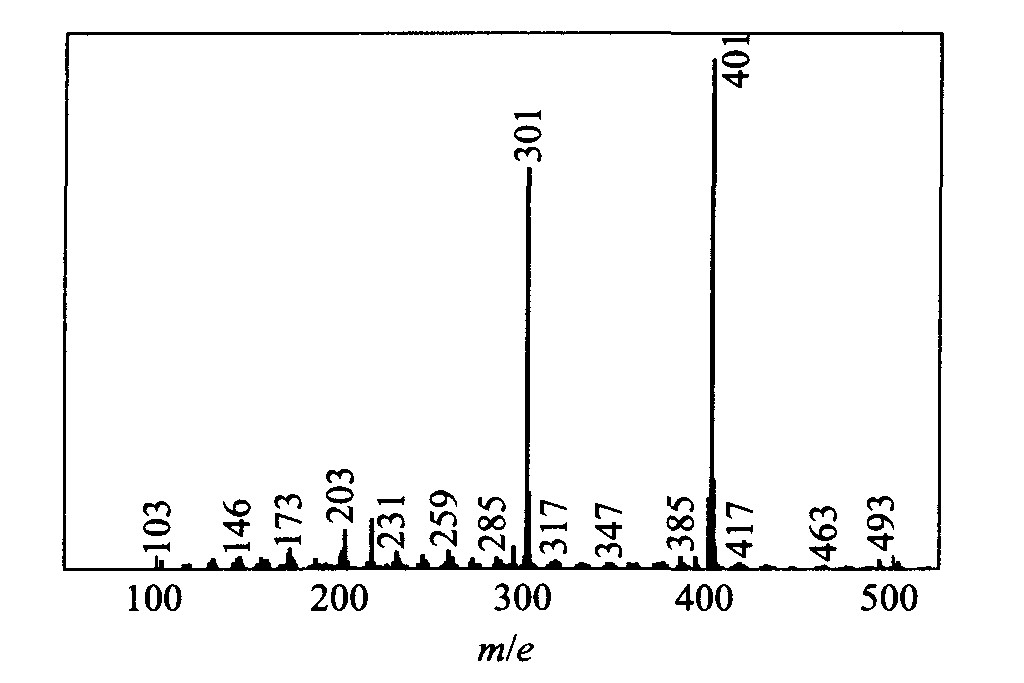
ͼ4 Rh(acac)3��FAB+����
Fig.4 FAB+ mass spectrum of Rh(acac)3
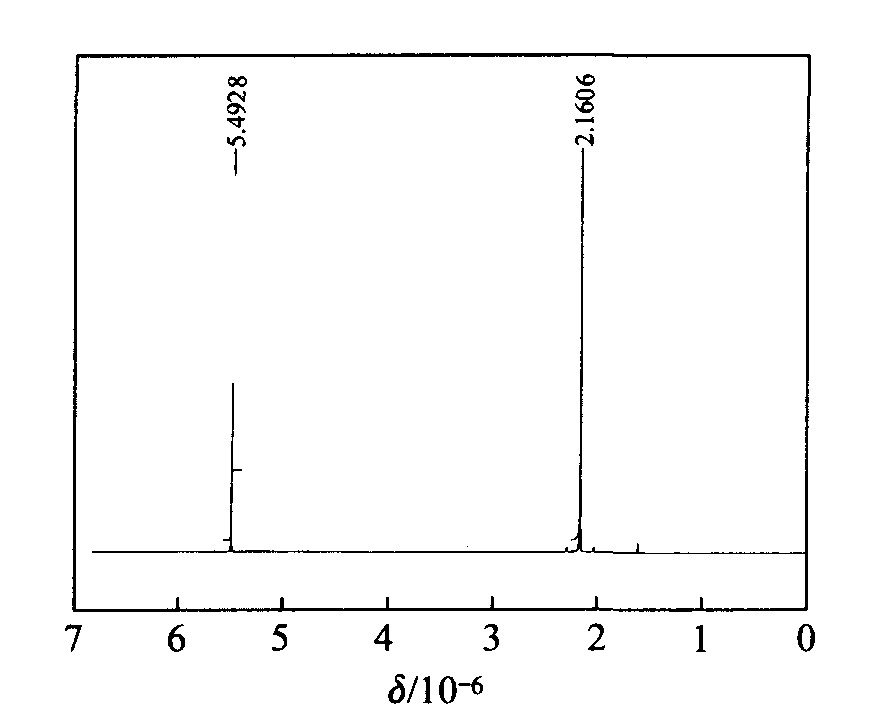
ͼ5 Rh(acac)3�ĺ˴Ź�������
Fig.51HNMR of Rh(acac)3
2.5 ������ͪ���TG-DTA����
ͼ6, 7��ʾ�ֱ�Ϊ������ͪ���ڿ������պ���������е�TG��DTA���ߡ�
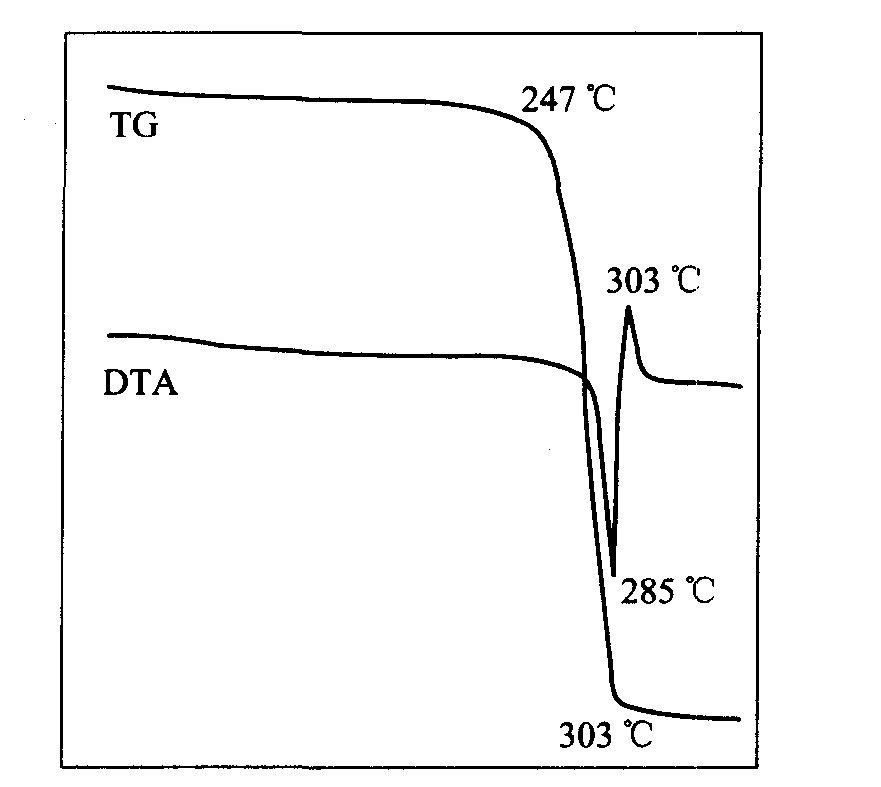
ͼ6 Rh(acac)3�ڿ����е�TG��DTA����
Fig.6 TG��DTA curves of Rh(acac)3 in air

ͼ7 Rh(acac)3������е�TG��DTA����
Fig.7 TG��DTA curves of Rh(acac)3 in argon
��ͼ6�е�TG���߿�֪, �ڿ���������, Rh(acac)3��������ʼ�¶�Ϊ247 ��, ��ʱ����ʼ��������, ��������ʧ, TG�����½�, ֱ��303 ��ʱ������ʧ��ֹ, ����������ƽ��, ˵�����������ȶ�������; ͬʱ�۲�������߷���, DTA�������¶����ߵ�ͬʱ����Խ��Խ���ҵ�����������ֽ�����, ��285 ��������ȷ嶥�� ͬʱ, Rh(acac)3�ֽ�����л������ڿ���������ȼ��, ȼ�շų���������DTA���ߵ������� �ֽ����ȷ������ �ڸDz������ɷ��ȷ�, ��303 ��ﵽ���ȷ嶥�¶�, �˺�Rh(acac)3�����غͲ������߶�����ƽ��, ��������ʣ�·ֽ�������ȶ��������, ������ʧ��Ϊ88.8%�� ����ѧ����ʽΪ
ʽ�� ��H��ʾ������
��ͼ7�е����������, Rh(acac)3��245 �濪ʼ������������, ��������ʧ, ͬʱ�����������ķֽ��Լ��ֽ��������л�������ٷֽ�, ����DTA���߳������Ե�ǿ���ȷ�, ����259 ��ﵽ���ȷ嶥�� �������������²�ͬ����, ������ͪ���ڶ��������²�������ȼ��, ֻ�������¶ȵ����߶��ӿ��������ѽ�, ������ַ��ȵĹ���, �����DTA������ֻ�������ȷ��û�з��ȷ塣 �����������¶�269 ��֮��, TG��DTA���߶�����ƽ��, ˵��Ҳ�������ȶ�������, ��������������ʧ��91.6%, �ɼ����ڶ������������������, ������ʧ����ȫ�� ����ѧ����ʽΪ:
2.6 GC-MS���������
Rh(acac)3�ڿ���������в�ͬ�¶��µ����ѽ����ֱ����ڱ�2�ͱ�3�С�
�ɱ�2���Կ���, ����������¶ȷ�Χ��, ����������, Rh(acac)3�ķֽ������Ҫ��������2,4-���ͪ�� ʵ����˵��: ���Ⱥ��ǿ��������λ�����ȶ�����������2,4-���ͪ��, ͬʱ�����˷�����������ԭ��Ӧ, ����ʧȥ����
��2 ����������Rh(acac)3���ȷֽ���Ҫ����
Table 2 Thermolytic products of Rh(acac)3 in air at different temperatures
Rh(acac)3 in air |
Percentage of peak area at different temperatures/% |
||||
250 �� |
300 �� | 400 �� | 500 �� | 600 �� | |
2,4-pentanedione |
82.70 | 96.91 | 93.7 | 92.3 | 79.50 |
Nonanal |
3.19 | 0.31 | 0.39 | 0.39 | 0.69 |
Benzyl benzoate |
2.19 | 0.38 | 0.21 | ||
3-acetyl-2,5-dimethyl furan |
0.66 | 0.39 | 1.53 | 0.28 | |
Rh(acac)3 |
10.67 | 0.35 | 4.62 | 2.53 | 2.43 |
��3 ���������Rh(acac)3���ȷֽ���Ҫ����
Table 3 Thermolytic products of Rh(acac)3 in argon at different temperatures
Product |
Percentage of peak area at different temperatures/% |
||||
250 �� |
300 �� | 400 �� | 500 �� | 600 �� | |
2,4-pentanedione |
50.28 | 62.96 | 53.15 | 52.55 | 46.66 |
1-propen-2-ol acetate |
34.56 | 35.38 | 42.57 | 40.76 | 40.33 |
3-buten-2-one |
2.10 | 3.80 | 5.68 | ||
3-penten-2-one |
0.28 | 0.18 | 1.58 | ||
2-butanone |
0.97 | 0.88 | 2.29 | ||
Furan |
0.55 | 0.42 | 0.63 | ||
Hexan-2,4-dione, enol |
0.37 | 1.78 | |||
����, �ڿ���������Rh(acac)3���ѽ������û�з��ֶ�����̼, ��һ��������л����ڿ������¶�Խ��Խ����ȼ��, Ҳ��Խ�������ɶ�����̼, ��ʵ����ȴ�������, �������ʵ�������йء� �����ѽ������, ���ѽ��������±��¹����ж����ܱյ�, ����������, ������������, ���ѽ�������ٲ��������ڸ����½���, �������ɻ���Ϸ�Ӧ, �����ѽ��Ҵ��ڸ��º�ȱ��״̬, ����ȼ�����ɵ�����������̼������ֽ������̼���ʷ�Ӧ����һ����̼, ��һ����̼�������ʺɱ�̫С�����ܱ�������; Ҳ���������������Ͳ���ȫ, ֻ�ܵ�����һ����̼�ĽΡ�
�ӱ�3�е����ݿ��Կ���: ����������, ����������¶ȷ�Χ��, 2,4-���ͪ��������Ҫ�ֽ����, ���京���Ͽ����������Ѵ�Ϊ����, ���������Ĵ��ڻ�����2,4-���ͪ�ѽ�����ɻ���Ƭ, ��������в���֪; �����¶ȵ����, ��Ƭ����Ϊ�ṹԽ��Խ����, ����Խ��Խ���������������, ��400 ���Rh(acac)3�ѽ�����Ҫ���ﻹ��������-2-��ϩ������ 3-��ϩ-2-ͪ�� 3-��ϩ-2-ͪ�� 2-��ͪ�� 4-�ǻ�-3-��ϩ-2-ͪ���, Ҳ�������Եؿ����¶ȶ�Rh(acac)3�����ѽ�̶ȵ�Ӱ�졣
���Կ���: ����������, ֻ����300 ��ʱ, ������ͪ��ֽ�������, 2,4-���ͪ���ȶ��Ļӷ��Բ��ﺬ����, ԭ��ʣ����С; ����������ͪ����Ϊǰ�����Ʊ���Ʋ�ʱ, ����¶ȿ��ԽϺõر������ڸ������л���ַֽ�������̼������
��Ȼ, CVD���չ��̵ij������ȡ� ���������ͳ������ʻ��ܵ���������ѧ��Ӧ�� ���涯����Ӧ�� ������ϼ����ȷ�ʽ��������Ӱ�졣 ���Ľ�����ǰ������Ȼ�ѧ�����ϵó����Ͻ���, Ϊ�����CVD����ղ����Ż��ṩ�ο���
3 ����
�����Ȼ���Ϊ��ʼԭ��, �ϳ���CVD�����Ʊ����ά���ϵ�ǰ����������ͪ��(��), ����Ϊ76%�� ͨ�����ӹ��ס� IR�� MS�� 1HNMR, �о���������ͪ��Ļ�ѧ�ṹ����, ������TG��DTA��GC��MS�о������ȷֽ���Ϊ�� �ṹ�����о�����: ������ͪ�����ڵ������Ķ��������, ����λȡ����Ӧ����ѧ�����ȶ���; ��IR��, ���������������м����γ�, �����ʻ��Ѳ��߱����͵��ʻ�����, Rh��O������λ��465.98 cm-1; �ڿ�ԭ�ӵĺ����, ������ͪ�����λ����һ����, �ͷų������������ͪ,����γ�103Rh+, ͬʱ�����Ź��ۼ��Ķ���; �ڴų���, ����Rh3+��ǿ������ЧӦ�Լ����ϻ��Ļ�������ЧӦ, H�˵����μ�С, ��ѧλ����ͳ��ƶ�, ʾ��������ͪ����������γ���λ���ϻ��������ǿ����λ��������Ҫԭ�� �ȷ����������: �ڿ��������������, ������ͪ��ֱ���247 ����245 ��ʱ��ʼ�����ֽⲢ����������ȼ�ա� �ֽ⼰���ɻ�����, ����ȼ���ͷŵ��������������ͷֽ����������; ������ͪ���ڿ���������е���Ҫ�ֽ���ﶼ��2,4-���ͪ, ���������������Ĵ���, ���ֲ����Ͷȸ���IJ���; 300 ��ʱ, ������ͪ��ֽ�������, 2,4-���ͪ���ȶ��Ļӷ��Բ��ﺬ����, ԭ��ʣ����С, ���ԽϺõر������ڸ������л���ַֽ�������̼������, ������ΪCVD����ij����¶ȡ�
�����
[1] ��HartleyFR.TheChemistryofPlatinumandPalladium[M].London:AppliedSciencePulishersLtd,1973.1012.
istry[M].Changsha:CentralSouthUniversityofTech nologyPress,1986.601604.
