
Trans. Nonferrous Met. Soc. China 30(2020) 1635-1646
Comparative genome analysis on intraspecific evolution and nitrogen fixation of Leptospirillum ferriphilum isolates
Hong-wei LIU1,2, Liang-feng XU1,2, Xue GUO1,2,3, Hui-dan JIANG4, Xue-duan LIU1,2, Yi-li LIANG1,2, Hua-qun YIN1,2, Ya-zi LIU1,2
1. School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Biometallurgy of Ministry of Education, Central South University, Changsha 410083, China;
3. State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100084, China;
4. Biotechnology Research Institute, Hunan Academy of Agricultural Sciences, Changsha 410125, China
Received 8 April 2019; accepted 28 April 2020
Abstract:
To reveal the intraspecific evolution of Leptospirillum ferriphilum isolates which thrived in industrial bioleaching ecosystems and acid mine drainages, genome sequences of L. ferriphilum YSK, L. ferriphilum DX and L. ferriphilum ZJ were determined to compare with complete genome of L. ferriphilum ML-04. The genome comparisons reveal that extensive intraspecific variation occurs in their genomes, and that the loss and insertion of novel gene blocks of probable phage origin may mostly contribute to heterogeneity of gene content among L. ferriphilum genomes. Surprisingly, a nif gene cluster is identified in L. ferriphilum YSK and L. ferriphilum ZJ genomes. Intensive analysis and further experiments indicate that the nif gene cluster in L. ferriphilum YSK inherits from ancestor rather than lateral gene transfer. Overall, results suggest that the population of L. ferriphilum undergoes frequent genetic recombination, resulting in many closely related genome types in recent evolution. The combinatorial processes profoundly shape their physiologies and provide the basis for adaptation to different niches.
Key words:
Leptospirillum ferriphilum; comparative genome; nitrogen fixation; intraspecific variation; recombination;
1 Introduction
Most chemolithotrophic iron- and sulfur- oxidizing bacteria and archaea can sustain by the oxidation of sulfide minerals, fixation of CO2 and N2 derived from air at extremely acidic environments (typically pH<3.0) such as acid mine drainage (AMD) [1,2]. These acidophiles are also widely used to recover metals, principally copper and gold, from low-grade ores and mineral concentrates via a process known as bioleaching or biomining [3,4]. However, variation and evolution of these microorganisms in extremely acidic environment remain poorly understood [1,5-7]. Recently, comparative genome analysis of individual species and metagenomic studies provide a first glimpse of genetic variation and population evolution of acidophiles that make these organisms survive in the extremely acidic niche [1]. Genome sequencing of eight strains of Thiomonas was carried out to evaluate genome variation, suggesting that the Thiomonas genome has evolved through the gain or loss of genomic islands and that the evolution is affected by the specific niches [8]. Metagenomic analysis of Leptospirillum dominated community in AMD demonstrated that significant intrapopulation sequence variation occurs due to recombination and mutation and to the acquisition or loss of gene features by phage, plasmid, or transposon insertion/deletion [6]. A comparison of the genome sequences of A. ferrooxidans ATCC 23270 and ATCC 53993 revealed that they exhibit 16% difference in gene content, although they are 100% identical at the ribosomal DNA level, and that these differences mainly result from lateral gene transfer [9]. However, little is known about the intraspecific genome variation and evolution of other extreme acidophiles.
As reported previously, the genus Leptospirillum, which is considered as dominant, moderately thermophilic microorganisms in industrial bioleaching ecosystems and acid mine drainages, can be divided into three groups, I, II and III, on the basis of 16S rRNA gene phylogeny [10,11]. L. ferrooxidans, L. ferriphilum and L. ferrodiazotrophum are the representatives of group I, II and III, respectively, and their genome sequences have been studied previously [12-15]. A whole nif gene cluster was found in the genomes of L. ferrooxidans and L. ferrodiazotrophum, but no nitrogenase genes were found in the genome of L. ferriphilum ML-04 and L. rubarum, two members of Leptospirillum group II, indicating that they cannot fix nitrogen [15]. Thus, it is inferred that Leptospirillum group II population lost nitrogen fixation ability after the speciation of Leptospirillum [12]. Because nitrogen resources (for example, ammonium, nitrate, and nitrite) are very limited in almost all AMD ecosystems [2,16], the loss of nitrogen fixation ability profoundly shaped the ecological niche differentiation of Leptospirillum group II population. The knowledge of intraspecific genome variation and evolution of Leptospirillum group II populations is critical to understand their ecological niche differentiation in these extremely acidic ecosystems.
In this study, we determined the complete genome sequence of L. ferriphilum YSK and draft genome sequences (100�� sequence coverage) of L. ferriphilum DX and L. ferriphilum ZJ, and comparative genome analysis between the three newly sequenced genomes and L. ferriphilum ML-04 genome was done. Surprisingly, a nif gene cluster was identified in both L. ferriphilum YSK and L. ferriphilum ZJ genomes, which was confirmed by physiological experiment and real- time quantitative polymerase chain reaction (PCR). Thus, further analysis on the origination of nif gene cluster in L. ferriphilum YSK was conducted.
2 Experimental
2.1 Growth of L. ferriphilum strains and DNA isolation
L. ferriphilum YSK was isolated from acid mine drainage of Dexing Copper Mine, China in 2007 and deposited in Central South University (Changsha, China) [17]. L. ferriphilum DX was isolated from chalcopyrite tailings of Dexing Copper Mine, China in 2013, and L. ferriphilum ZJ was isolated from chalcopyrite tailings of Zijinshang, southeast China in 2013. They were grown at 40 ��C aerobically in 9K basal salt medium [18] with 4.5% (w/v) ferrous sulfate, separately. The 9K basal salt medium contained 3 g/L (NH4)2SO4, 0.1 g/L KCl, 0.5 g/L K2HPO4, 0.5 g/L MgSO4��7H2O and 0.01 g/L Ca(NO3)2. The pH of medium was adjusted to be 1.6, and then the medium was autoclaved for 25 min at 121 ��C. Microbial cells were harvested by centrifugation (15000g) for 10 min at 4 ��C. Genomic DNA was extracted from the collected cells using TIANamp Bacteria DNA kit (TIANGEN, China) according to the manufacturer��s instruction and finally suspended in MilliQ water and stored at -80 ��C until used for genome sequencing.
2.2 Genome sequencing, assembly and annotation
Genomic library construction, sequencing, and assembly of L. ferriphilum YSK, L. ferriphilum ZJ and L. ferriphilum DX were performed at the Beijing Genomics Institute (BGI; Shenzhen, China) by using SOAPdenovo package [19,20]. To obtain the complete genome sequence of L. ferriphilum YSK, sequencing was further assembled by using the Ion Torrent PGM platform, Newbler assembler and sequencing gap-spanning PCR [21]. Manual editing of genome sequence was done using the DNAstar software package (Madison, WI). All coding sequences (CDSs) were predicted by using Glimmer 3.02 [22], and manually curated and verified by using the annotation software BLAST [23-26]. The unassigned CDSs were further annotated using the hmmpfam program of the HMMER package [27]. Furthermore, transporter gene annotations were performed by additionally taking into account the information of transporter classification database [28]. Transfer RNA (tRNA) genes were predicted by using tRNAscan-SE [29], and the ribosomal RNA (rRNA) genes were identified by RNAmmer program [30]. The insertion sequences (IS) were searched by using the IS Finder (https://www-is.biotoul.fr/), and the annotated transposons were searched against NCBI nucleotide sequence databases to find IS elements. The genomic sequences reported in this work have been deposited in the NCBI GenBank database under accession numbers PRJNA232614 (YSK), PRJNA244732 (DX), PRJNA244731 (ZJ).
2.3 Comparative genome analysis
The genome sequences of L. ferriphilum YSK and L. ferriphilum ML-04 were compared pairwise using the Artemis Comparison Tool (ACT) [31]. Orthologous gene sets between them were identified by reciprocal FASTA searches. Only those pairs of homologous gene were selected for further analysis where the amino acid identity ��50% over 80% of the protein length. These genes were subject to manual curation using gene synteny to increase the accuracy. Laterally transferred genomic regions were identified by using Alien_hunter [32]. Visualization of genome was performed with Circos [33]. Furthermore, the GGDC-Genome-to-Genome Distance Calculator was used to estimate the overall similarity among these isolate genomes [34]. The system calculates the distances by comparing the genomes to obtain HSPs (high-scoring segment pairs) and interfering distances from three formulae (HSP length/total length; identities/HSP length; identities/total length) [35]. The inferred digital DNA-DNA hybridization values were calculated.
2.4 Phylogenetic analysis
Phylogenetic analysis of 16S rRNA and nifH genes was performed on sequences from L. ferriphilum isolate genomes and reference sequences in the NCBI GenBank database. These 16S rRNA gene sequences were aligned with the multiple sequence alignment tool ClustalW 2.0 [36], and phylogenetic trees were constructed by using MEGA, version 4.0 [37]. Deduced amino acid sequences for nifH genes of L. ferriphilum isolate genomes were aligned with known nifH sequences from the NCBI GenBank database with ClustalW 2.0.
2.5 Nitrogen-free growth and RNA extraction
The nitrogen-free 9K medium was modified by removal of Ca(NO3)2 and (NH4)2SO4, the sources of nitrogen in the medium. L. ferriphilum YSK was grown in nitrogen-free 9K medium and standard 9K medium at 40 ��C with FeSO4��7H2O as the primary energy source. Microbial cells were harvested and total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, USA), treated with RNase-free DNase I (Qiagen, Valencia, USA) and purified with an RNeasy kit (Qiagen, Valencia, USA). RNA concentration and purity were measured with a NanoDrop 2000 spectrophoto- meter (ThermoScientific), and then stored at -80 ��C until used for real-time quantitative PCR analysis.
2.6 Real-time quantitative PCR
The specific primers were designed for nifA, nifH, nirK, nirA, amtB and 16S rRNA genes by Primer Premier 5.0 (Table 1).
Table 1 Specific primers used in real-time quantitative PCR
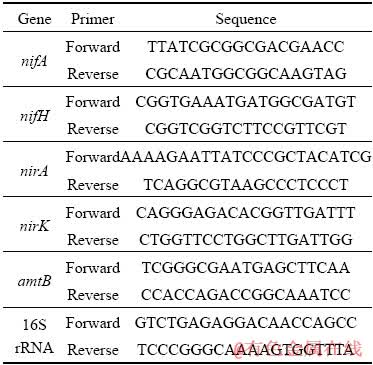
Purified RNA was used for single-stranded cDNA synthesis with the ReverTra Ace qPCR RT Kit (TOYOBO). Real-time quantitative PCR in three duplicates was performed with iCycler iQ Real-Time PCR detection system (Bio-Rad Laboratories Inc., Hercules, USA). 25 ��L reaction mixture contained 12.5 ��L SYBROR Green Real- Time PCR Master Mix (Toyobo Co., Ltd., Osaka, Japan), 0.5 ��L template single-stranded cDNA, and 1 ��L each of 10 ��mol/L forward and reverse primers, and 10 ��L deionized water. The specific amplification protocol was as follows: 95 ��C for 5 min, then 40 cycles of 95 ��C for 20 s, 55 ��C for 15 s, and 72 ��C for 15 s and final incubation of 72 ��C for 10 min. 16S rRNA gene was used as the reference gene for normalization. The relative expression of target genes was calculated using comparative DDCT method [38].
3 Results and discussion
3.1 General features of genomes of L. ferriphilum isolates
The draft genomes of L. ferriphilum YSK, L. ferriphilum DX and L. ferriphilum ZJ were sequenced and annotated. Furthermore, the complete genome sequence of L. ferriphilum YSK was determined by finishing the gaps between contigs. The general genome features of L. ferriphilum YSK, L. ferriphilum DX and L. ferriphilum ZJ are shown in Table 2.
Compared to the complete genome of L. ferriphilum ML-04 [13], three newly-sequenced genomes of L. ferriphilum isolates have smaller sizes and the similar GC content (approximately 54%) (Table 2). The GGDC-Genome-to-Genome Distance Calculator revealed that the pairwise genome similarity of three newly-sequenced isolates and L. ferriphilum ML-04 is more than 90%, suggesting that the similarity and synteny of core regions are still predominant among L. ferriphilum isolate genomes. Furthermore, 16s rRNA gene sequences of four L. ferriphilum isolates share more than 99% nucleotide identity and a monophyletic group clustering forms within other Leptospirillum species in phylogenetic tree (Fig. 1).
However, many different features are found in the genomes of the four L. ferriphilum isolates. The most striking feature is numerous genes coding for transposases/integrase scattered across all L. ferriphilum isolate genomes (Table 2). Among the four isolate genomes, L. ferriphilum ML-04 and L. ferriphilum DX have rather more transposases/ integrase genes and tandem repeats than L. ferriphilum YSK and L. ferriphilum ZJ. These characteristics of L. ferriphilum isolate may provide clues of genomic heterogeneity [39]. Previous metagenomic analysis of Leptospirillum dominated community in AMD also revealed that intrapopulation sequence variation occurring in Leptospirillum genera mainly results from recombination, mutation and the acquisition or loss of gene features by phage, plasmid, or transposon insertion/deletion [6]. In addition, two 5S-16S-23S rRNA operons are identified in the complete genomes of L. ferriphilum YSK and L. ferriphilum ML-04, but only one 5S-16S-23S rRNA operon is found in the draft genomes of L. ferriphilum DX and L. ferriphilum ZJ, which may lose the other 5S-16S-23S rRNA operon in the assembly of draft genomes (Table 2). The genome heterogeneity among islolates of L. ferriphilum results in differential gene contents including some physiolo- gically important genes. For example, comparative genome analysis highlights an important hetero- geneous region including a nif gene cluster among these genomes. A nif gene cluster including structural subunit genes nifH, nifD, nifK and various additional subunit genes is found in L. ferriphilum YSK and L. ferriphilum ZJ, while no nif gene cluster is identified in L. ferriphilum DX and L. ferriphilum ML-04 (Table 2).
Table 2 General features of L. ferriphilum isolate genomes


Fig. 1 Phylogenetic analysis result of 16S rRNA genes
3.2 Whole genome comparison of isolate YSK and ML-04
To obtain more accurate insights into intraspecific genome variation and evolution of L. ferriphilum isolates, genome of L. ferriphilum YSK was compared with the genome of L. ferriphilum ML-04, a well-characterized and fully-sequenced L. ferriphilum isolate [13]. An alignment of the genome of L. ferriphilum YSK with that of L. ferriphilum ML-04 reveals numerous deletion and/or insertion genomic blocks (Fig. 2).
In Fig. 2, from the outside in, the outer Circle 1 marks the size in base pairs. Circles 2 and 3 show the position of CDSs transcribed in a clockwise and anti-clockwise direction, respectively. Circles 4 and 5 show the positions of genomic regions ofL. ferriphilum YSK that lacks orthologs in L. ferriphilum ML-04 and the positions of genomic regions of L. ferriphilum ML-04 that lacks orthologs in L. ferriphilum YSK, respectively. Circles 6 and 7 show unique ortholog genes in L. ferriphilum YSK and L. ferriphilum ML-04, respectively. Circles 8 and 9 show the positions of transposase genes in L. ferriphilum YSK and L. ferriphilum ML-04, respectively. Circle 10 shows a plot of GC skew ([G-C]/[G+C]), in a 1 kb window). Genes in Circles 2 and 3 are color-coded according to the function of their gene products: dark green, membrane or surface structures; yellow, central or intermediary metabolism; cyan, degradation of macromolecules; red, information transfer/cell division; cerise, degradation of small molecules; pale blue, regulators; salmon pink, pathogenicity or adaptation; black, energy metabolism; orange, conserved hypothetical; pale green, unknown; and brown, pseudogenes.
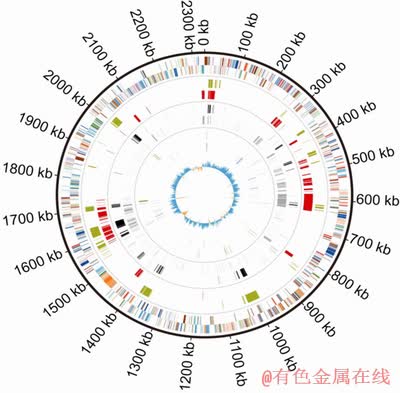
Fig. 2 Circular representation of L. ferriphilum YSK genome
Although the two isolate genomes share 2012 CDSs with >90% average nucleotide identity, the genes that are only present either in L. ferriphilum YSK or L. ferriphilum ML-04 form 10.97% and 18.58% of their respective genomes (Fig. 2 and Fig. 3).
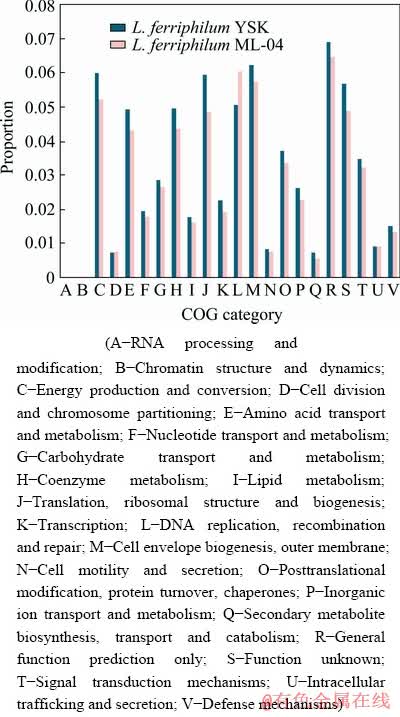
Fig. 3 Comparison of proportions of functional categories in genomes of L. ferriphilum YSK and L. ferriphilum ML-04
Interestingly, most of unique CDSs in L. ferriphilum YSK and L. ferriphilum ML-04 are in clusters from >3 kb up to >39 kb in deletion and/or insertion of genomic blocks, and there are few indels of <3 kb (Fig. 2). Furthermore, these variant genomic blocks predominantly encode hypothetical proteins, as well as transposases, integrases, or proteins involved in plasmid replication and/or transmission (Fig. 2). The results indicate that these variant genomic blocks may be mediated by integrated elements of putative phage, plasmid, or transposon origin. Interestingly, some distinct genomic blocks encode physiologically important genes involved in core energy metabolism, cofactor biosynthesis and transcription. For example, a gene encoding sulfate adenylyltransferase in an insertion of genomic block is identified in L. ferriphilum YSK. Sulfate adenylyltransferase is a key enzyme in sulfur metabolism, which is required for PAPS (phosphoadenosine-phosphosulfate) synthesis from inorganic sulfate [40]. Comparative COG analysis also reveals that the majority of L. ferriphilum YSK unique CDSs belong to the COGs categories of energy production and conversion, amino acid transport and metabolism, and coenzyme metabolism (Fig. 3).
Genomic heterogeneity between L. ferriphilum YSK and L. ferriphilum ML-04 arises from localized variation in gene content, typically focused in the integrated plasmid/phage-like regions. Movement of insertion sequence (IS) elements can give rise to strain-level differences. In both L. ferriphilum YSK and L. ferriphilum ML-04, distinct genomic regions are often associated with transposase or phage-type integrase genes (Fig. 2). There are 38 transposase/integrase insertions in L. ferriphilum YSK and 53 in L. ferriphilum ML-04. However, more than 90% transposase/integrase genes are found in different genomic regions between the genomes of L. ferriphilum YSK and L. ferriphilum ML-04 (Fig. 2). There are very few transposases occurring in the same location of L. ferriphilum YSK and L. ferriphilum ML-04 genomes (Fig. 2). These results indicate that transposon movement and the loss and insertions of gene blocks of probable phage origin may mostly contribute to genomic heterogeneity of L. ferriphilum isolates.
3.3 nif gene cluster in L. ferriphillum YSK
A complete nif operon including nifH, nifD and nifK was identified in a 31-kbp insertion of genomic block in L. ferriphillum YSK genome (Fig. 4).
In Fig. 4, a nif gene cluster is only present in YSK and distinct from the comparable region in ML-04. The 40-gene insertion in YSK (shown in red) includes nitrogenase structural subunits nifHDK, numerous additional subunits, nitrogen regulator, molybdenum transporter and numerous conserved hypothetical proteins. These genes are also shown in Table 2.
The nifH, nifD and nifK genes, coding for a structural subunit of dinitrogenase reductase and two Fe-Mo subunits of dinitrogenase, respectively, are required for the functional nitrogenase in almost all diazotrophs [2]. In many nitrogen-fixing microorganisms such as Azotobacter vinelandii, Herbaspirillum speropedicae and Bradyrhizobium japonicum, these nitrogenases have similar sequences and common structures [41-43]. Phylogenetic analysis of nifH proteins indicates that for L. ferriphillum YSK a monophyletic group clustering forms within other Leptospirillum species (Fig. 5).
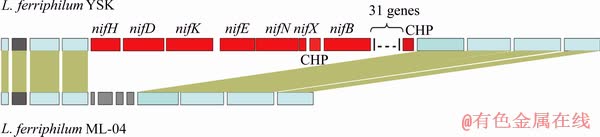
Fig. 4 Heteromorphic genomic region between L. ferriphilum YSK and L. ferriphilum ML-04

Fig. 5 Phylogenetic tree inferred from nifH proteins
Furthermore, numerous additional nif subunit genes including nifE, nifN, nifX, nifB, nifS, nifZ, nifT and nifW are also identified in this nif gene cluster (Fig. 4 and Table 3), which play important roles in the regulation of nif genes and maturation processes of inactive products, such as electron transport and FeMo-cofactor biosynthesis and assembly [42]. And these additional nif subunits are most similar with the gene products of either L. ferrooxidans or L. ferrodiazotrophum (Table 3). These results indicate that the nif gene cluster of L. ferriphillum YSK has a high level of phylogenetic homology with members of Leptospirillum groups I and III. Therefore, it is inferred that the nif gene cluster in L. ferriphilum YSK is obtained via inheritance from ancestor rather than lateral gene transfer.
Table 3 Gene products in heteromorphic genomic region

Comparative genome analysis reveals that although no nif gene cluster is found in the complete genome of L. ferriphillum ML-04 [13], the surrounding genomic context of the nif gene cluster in L. ferriphillum YSK displays sequence similarities and synteny with that of L. ferriphillum ML-04 (Fig. 4). Furthermore, a very similar nif gene cluster is identified in L. ferriphillum ZJ but not in L. ferriphillum DX (Table 2). These results indicate that not all strains of Leptospirillum group II have lost the nif gene cluster, suggesting that the loss of nif gene cluster may occur in the very recent evolutionary period of Leptospirillum group II rather than in the Leptospirillum speciation. Interestingly, the genomes of L. ferriphillum ML-04 and L. ferriphillum DX that have lost the nif gene cluster have more transposons and tandem repeats than the genomes of L. ferriphillum YSK and L. ferriphillum ZJ that have a nif gene cluster (Table 2). Normally, inter-specific and intra-specific transposon movements can significantly increase the heterogeneity of genomes [6,44]. It is highly possible that some of Leptospirillum group II members lose the nif gene cluster through transposon movements.
3.4 Quantitative N-cycling gene expression
To determine the nitrogen fixation ability of L. ferriphilum YSK, L. ferriphilum YSK was grown in 9K medium both in the presence and in the absence of fixed nitrogen. The results of physiological experiment show that the medium changes from pale green to a deep-red color more quickly in the nitrogen-free media, indicating faster iron oxidation, possibly due to the increased need for energy for nitrogen fixation, and the similar results have been observed in the culture of L. ferrooxidans in the nitrogen-free media in a previous study [12]. The key gene expressions of nitrogen metabolism in L. ferriphilum YSK in the nitrogen-free media were determined by using real-time quantitative PCR (qRT-PCR) (Fig. 6).
The expressions of these nitrogen-related genes [45] show remarkable up-regulations in the nitrogen-free media. The obvious up-regulation of nifA gene is observed in the nitrogen-free media, indicating that the transcription of nif gene operon in L. ferriphilum YSK may be up-regulated. Consistently, the key nitrogen-fixing gene, nifH, has a 2-fold higher expression level in the nitrogen-free media than in control media, indicating that the nif gene operon in L. ferriphilum YSK is indeed activated and that the nitrogen fixation is significantly enhanced in the nitrogen-free media. Furthermore, nirK coding for nitrite reductase and amtB coding for ammonium transporter, which play important roles in denitrification and ammonium transportation, respectively, also exhibit up-regulated expression levels in the nitrogen-free media (Fig. 6). These results indicate that not only nitrogen fixation but also some of other nitrogen cycling processes in L. ferriphilum YSK are significantly enhanced to respond to the lack of fixed nitrogen.
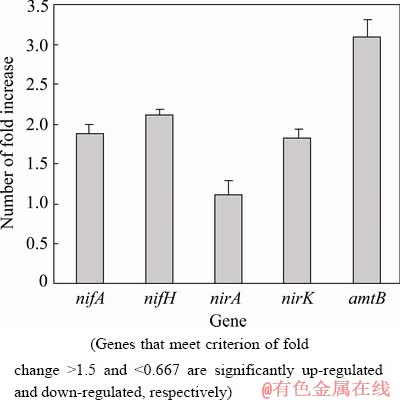
Fig. 6 Expression changes of key nitrogen cycling genes in nitrogen-free media
4 Conclusions
(1) Comparative genome analysis of the three newly-sequenced genomes and L. ferriphilum ML-04 genome shows that extensive intraspecific variations occur in these L. ferriphilum isolate genomes, and that transposon movements and the loss and insertion of gene blocks of probable phage origin may mostly contribute to the genome heterogeneity among L. ferriphilum isolates.
(2) A whole nif gene cluster is identified in both L. ferriphilum YSK and L. ferriphilum ZJ genomes, indicating that the nif gene cluster is obtained via inheritance from ancestor rather than lateral gene transfer, and that the loss of nif gene cluster may occur in the very recent evolutionary period of Leptospirillum group II rather than in the Leptospirillum speciation.
(3) Physiological experiment and real-time quantitative PCR further confirm that L. ferriphilum YSK indeed has the nitrogen fixation ability. It is suggested that intraspecific genome variations among L. ferriphilum genomes profoundly shape their physiologies and provide the basis for adaptation to different niches.
References
[1] CArdenas JP, ValdEs J, Quatrini R, Duarte F, Holmes DS. Lessons from the genomes of extremely acidophilic bacteria and archaea with special emphasis on bioleaching microorganisms [J]. Applied Microbiology and Biotechnology, 2010, 88: 605-620.
[2] Baker B J, Banfield J F. Microbial communities in acid mine drainage [J]. FEMS Microbiology Ecology, 2003, 44: 139-152.
[3] Rawlings D E, Johnson D B. The microbiology of biomining: Development and optimization of mineral- oxidizing microbial consortia [J]. Microbiology, 2007, 153: 315-324.
[4] ZHU Ping, LIU Xue-duan, CHEN Ai-jia, LIU Hong-wei, YIN Hua-qun, QIU Guan-zhou, HAO Xiao-dong, Liang Yi-li. Comparative study on chalcopyrite bioleaching with assistance of different carbon materials by mixed moderate thermophiles [J]. Transactions of Nonferrous Metals Society of China, 2019, 29: 1294-1303.
[5] Hao Xiao-dong, Liu Xue-duan, Yang Qin, Liu Hong-wei, Yin Hua-qun, Qiu Guan-zhou, Liang Yi-li. Comparative study on bioleaching of two different types of low-grade copper tailings by mixed moderate thermophiles [J]. Transactions of Nonferrous Metals Society of China, 2018, 28: 1847-1853.
[6] Simmons S L, DiBartolo G, Denef V J, Goltsman D S A, Thelen M P, Banfield J F. Population genomic analysis of strain variation in Leptospirillum Group II bacteria involved in acid mine drainage formation [J]. PLOS Biology, 2008, 6: e177. DOI: 10.1371/journal.pbio.0060177.
[7] Vald��s J, C��rdenas J P, Quatrini R, Esparza M, Osorio H, Duarte F, Lefimil C, Sepulveda R, Jedlicki E, Holmes D S. Comparative genomics begins to unravel the ecophysiology of bioleaching [J]. Hydrometallurgy, 2010, 104: 471-476.
[8] Ars��ne-Ploetze F, Koechler S, Marchal M, Copp��e J Y, Chandler M, Bonnefoy V, Brochier-Armanet C, Barakat M, Barbe V, Battaglia-Brunet F. Structure, function, and evolution of the Thiomonas spp. genome [J]. PLoS Genet, 2010, 6: e1000859-e1000859.
[9] Levic��n G, Katz A, Vald��s J H, Quatrini R, Holmes D S, Orellana O. A 300 kpb genome segment, including a complete set of tRNA genes, is dispensable for Acidithiobacillus ferrooxidans [M]. Switzerland: Trans Tech Publ. 2006, 187-190.
[10] Hippe H. Leptospirillum gen. nov. (ex Markosyan 1972), nom. rev., including Leptospirillum ferrooxidans sp. nov. (ex Markosyan 1972), nom. rev. and Leptospirillum thermoferrooxidans sp. nov. (Golovacheva et al. 1992) [J]. International Journal of Systematic and Evolutionary Microbiology, 2000, 50: 501-503.
[11] PENG Tang-jian, ZHOU Dan, LIU Ya-nan, YU Run-lan, QIU Guan-zhou, ZENG Wei-min. Effects of pH value on the expression of key iron/sulfur oxidation genes during bioleaching of chalcopyrite on thermophilic condition [J]. Annals of Microbiology, 2019, 69: 627-635. https://doi. org/10.1007/ s13213-019-01453-y.
[12] Tyson G W, Lo I, Baker B J, Allen E E, Hugenholtz P, Banfield J F. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community [J]. Applied and Environmental Microbiology, 2005, 71: 6319-6324.
[13] Mi S, Song J, Lin J, Che Y, Zheng H, Lin J. Complete genome of Leptospirillum ferriphilum ML-04 provides insight into its physiology and environmental adaptation [J]. The Journal of Microbiology, 2011, 49: 890-901.
[14] Fujimura R, Sato Y, Nishizawa T, Oshima K, Kim S W, Hattori M, Kamijo T, Ohta H. Complete genome sequence of Leptospirillum ferrooxidans strain C2-3, isolated from a fresh volcanic ash deposit on the island of Miyake, Japan [J]. Journal of Bacteriology, 2012, 194: 4122-4123.
[15] Goltsman D S A, Denef V J, Singer S W, VerBerkmoes N C, Lefsrud M, Mueller R S, Dick G J, Sun C L, Wheeler K E, Zemla A. Community genomic and proteomic analyses of chemoautotrophic iron-oxidizing ��Leptospirillum rubarum�� (Group II) and ��Leptospirillum ferrodiazotrophum�� (Group III) bacteria in acid mine drainage biofilms [J]. Applied and Environmental Microbiology, 2009, 75: 4599-4615.
[16] Chen L x, Hu M, Huang L n, Hua Z s, Kuang J l, Li S j, Shu W s. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage [J]. ISME Journal, 2015, 9: 1579-1592.
[17] GAO J, ZHANG C G, WU X L, WANG H H, QIU G Z. Isolation and identification of a strain ofLeptospirillum ferriphilum from an extreme acid mine drainage site [J]. Annals of Microbiology, 2007, 57: 171-176.
[18] Silverman M P, Lundgren D G. Studies on the chemoautotrophic iron bacterium Ferrobacillus ferrooxidans I: An improved medium and a harvesting procedure for securing high cell yields [J]. Journal of Bacteriology, 1959, 77: 642-647.
[19] Minoche A E, Dohm J C, Himmelbauer H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems [J]. Genome Biology, 2011, 12: R112. URL: https://link. springer.com/article/10.1186/gb-2011-12-11-r112.
[20] Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y. SOAP denovo2: An empirically improved memory-efficient short-read de novo assembler [M]. Oxford: Gigascience, 2012: 18. https://link.springer. com/article/10.1186/2047-217X-1-18.
[21] Chaisson M J, Pevzner P A. Short read fragment assembly of bacterial genomes [J]. Genome Research, 2008, 18: 324-330.
[22] Delcher A L, Bratke K A, Powers E C, Salzberg S L. Identifying bacterial genes and endosymbiont DNA with Glimmer [J]. Bioinformatics, 2007, 23: 673-679.
[23] Sayers E W, Barrett T, Benson D A, Bolton E, Bryant S H, Canese K, Chetvernin V, Church D M, DiCuccio M, Federhen S. Database resources of the national center for biotechnology information [J]. Nucleic Acids Research, 2011, 39: D38-D51.
[24] Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes [J]. Nucleic Acids Research, 2000, 28: 27-30.
[25] Tatusov R L, Fedorova N D, Jackson J D, Jacobs A R, Kiryutin B, Koonin E V, Krylov D M, Mazumder R, Mekhedov S L, Nikolskaya A N. The COG database: An updated version includes eukaryotes [J]. BMC Bioinformatics, 2003, 4: 41-55.
[26] Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden T L. NCBI BLAST: A better web interface [J]. Nucleic Acids Research, 2008, 36: W5-W9.
[27] Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J. The Pfam protein families database [J]. Nucleic Acids Research, 2012, 40: D290-D301.
[28] Saier M H, Tran C V, Barabote R D. TCDB: The transporter classification database for membrane transport protein analyses and information [J]. Nucleic Acids Research, 2006, 34: D181-D186.
[29] Lowe T M, Eddy S R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence [J]. Nucleic Acids Research, 1997, 25: 955-964.
[30] Lagesen K, Hallin P, Rodland E A, Starfeldt H H, Rognes T, Ussery D W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes [J]. Nucleic Acids Research, 2007, 35: 3100-3108.
[31] Carver T J, Rutherford K M, Berriman M, Rajandream M-A, Barrell B G, Parkhill J. ACT: The Artemis comparison tool [J]. Bioinformatics, 2005, 21: 3422-3423.
[32] Vernikos G S, Parkhill J. Interpolated variable order motifs for identification of horizontally acquired DNA: Revisiting the Salmonella pathogenicity islands [J]. Bioinformatics, 2006, 22: 2196-2203.
[33] Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones S J, Marra M A. Circos: An information aesthetic for comparative genomics [J]. Genome Research, 2009, 19:1639-1645.
[34] Meier-Kolthoff J P, Auch A F, Klenk H P, Goker M. Genome sequence-based species delimitation with confidence intervals and improved distance functions [J]. BMC Bioinformatics, 2013, 14: 60-74.
[35] Auch A F, von Jan M, Klenk H P, Goker M. Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison [J]. Standards in Genomic Sciences, 2010, 2: 117-134.
[36] Thompson J D, Gibson T, Higgins D G. Multiple sequence alignment using ClustalW and ClustalX [M]. Hoboken: Current Protocols in Bioinformatics, 2002: 2.3. 1-2.3. 22.
[37] Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0 [J]. Molecular Biology and Evolution, 2007, 24: 1596-1599.
[38] Cikos S, Bukovska A, Koppel J. Relative quantification of mRNA: Comparison of methods currently used for real-time PCR data analysisv [J]. BMC Molecular Biology, 2007, 8: 113-127.
[39] KAZAZIAN H H. Mobile elements: Drivers of genome evolution [J]. Science, 2004, 303: 1626-1632.
[40] Kurima K, Warman M L, Krishnan S, Domowicz M, Krueger R C, Deyrup A, Schwartz N B. A member of a family of sulfate-activating enzymes causes murine brachymorphism [J]. Proceedings of the National Academy of Sciences, 1998, 95: 8681-8685.
[41] Bond P L, Smriga S P, Banfield J F. Phylogeny of microorganisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site [J]. Applied and Environmental Microbiology, 2000, 66: 3842-3849.
[42] Dai Z M, Guo X, Yin H Q, Liang Y L, Cong J, Liu X D. Identification of nitrogen-fixing genes and gene clusters from metagenomic library of acid mine drainage [J]. PLOS ONE, 2014, 9: e87976. https://journals.plos.org/plosone/ article?id=10.1371/journal.pone.0087976.
[43] Kallas T, Coursin T, Rippka R. Different organization of nif genes in nonheterocystous and heterocystous cyanobacteria [J]. Plant Molecular Biology, 1985, 5: 321-329.
[44] Allen E E, Tyson G W, Whitaker R J, Detter J C, Richardson P M, Banfield J F. Genome dynamics in a natural archaeal population [J]. Proceedings of the National Academy of Sciences, 2007, 104: 1883-1888.
[45] Leigh J A, Dodsworth J A. Nitrogen regulation in bacteria and archaea [J]. Annual Review of Microbiology, 2007, 61: 349-377.
�������������������ڽ����̵��ıȽϻ��������
����ΰ1,2��������1,2���� ѩ1,2,3�����۵�4����ѧ��1,2�������� 1,2������Ⱥ1,2��������1,2
1. ���ϴ�ѧ ��Դ�ӹ������﹤��ѧԺ����ɳ 410083��
2. ���ϴ�ѧ ����ұ��������ص�ʵ���ң���ɳ 410083��
3. �廪��ѧ ����ѧԺ������ģ������Ⱦ���ƹ����ص�����ʵ���ң����� 100084;
4. ����ʡũҵ��ѧԺ ũҵ���\���о�������ɳ 410125
ժ Ҫ��Ϊ��ʾ������ұ��ҵ��̬ϵͳ�����Կ�ɽ��ˮϵͳ��ռ������λ����������������(Leptospirillum ferriphilum)���ڽ������ƣ�������������������L. ferriphilum YSK��L. ferriphilum DX ��L. ferriphilum ZJ��ȫ����������Ϊ�о�����ͨ����ͨ�����������е�L. ferriphilum ML-04����ȫ���������н��бȽϻ�����ѧ�о�������������������������������㷺�����ڱ��죬������Դ���ɾ�����»���Ƭ�ε�ȱʧ������ǵ���������������������������������Ե���Ҫԭ�����⣬����ط�������������������L. ferriphilum YSK �� L. ferriphilum ZJ�Ļ������д��ڹ̵�����ء������������һ��ʵ��֤ʵ����L. ferriphilum YSK�Ĺ̵�����ؼ̳�����ԭʼ���ȶ���Դ�Ժ������ת�ơ��ۺ������о������֪������������������Ⱥ��������ұ����ϵ�о���Ƶ���Ļ������飬�ڽ��ڵĽ����в������������صĻ��������ͣ�ͬʱ�����������������������������������������ԣ���Ϊ����Ӧ��ͬ����̬λ�춨������
�ؼ��ʣ������������������Ƚϻ����飻�̵������ڱ��죻��������
(Edited by Bing YANG)
Foundation item: Project (2018YFC1801804) supported by the National Key R&D Program of China; Projects (2016JJ3146, 2017JJ3160) supported by the Natural Science Foundation of Hunan Province, China
Corresponding author: Ya-zi LIU; Tel: +86-731-88830546; E-mail: 1579383816@qq.com
DOI: 10.1016/S1003-6326(20)65326-2
Abstract: To reveal the intraspecific evolution of Leptospirillum ferriphilum isolates which thrived in industrial bioleaching ecosystems and acid mine drainages, genome sequences of L. ferriphilum YSK, L. ferriphilum DX and L. ferriphilum ZJ were determined to compare with complete genome of L. ferriphilum ML-04. The genome comparisons reveal that extensive intraspecific variation occurs in their genomes, and that the loss and insertion of novel gene blocks of probable phage origin may mostly contribute to heterogeneity of gene content among L. ferriphilum genomes. Surprisingly, a nif gene cluster is identified in L. ferriphilum YSK and L. ferriphilum ZJ genomes. Intensive analysis and further experiments indicate that the nif gene cluster in L. ferriphilum YSK inherits from ancestor rather than lateral gene transfer. Overall, results suggest that the population of L. ferriphilum undergoes frequent genetic recombination, resulting in many closely related genome types in recent evolution. The combinatorial processes profoundly shape their physiologies and provide the basis for adaptation to different niches.
[39] KAZAZIAN H H. Mobile elements: Drivers of genome evolution [J]. Science, 2004, 303: 1626-1632.
" target="blank">[45] Leigh J A, Dodsworth J A. Nitrogen regulation in bacteria and archaea [J]. Annual Review of Microbiology, 2007, 61: 349-377.
