
�����������⻯��LaNi5Hx�ĵ�һ��ԭ��
�Ŵ��1, �� ��1, ���»�1, �� ��1, ���ƹ�1, ������1, �� ��2
( 1. �Ĵ���ѧ ԭ�ӷ��������о���, �ɶ� 610065;
2. �й����������о�Ժ �����뻯ѧ�о���, ���� 621900)
ժ Ҫ��
���ܶȷ�������Ϊ�����ĵ�һ��ԭ������, ����ȫ����������ƽ�沨����, ��LaNi5Hx(x=3, 4, 5)�����˽�ģ�о�, �����Ż��õ��ȶ��ľ���ṹ, ����������֤�������, ��������������ǵĵ����ܶ��Լ�״̬�ܶ�ͼ�� �������: �ڼ���ģ����, Hԭ�������ȶ����Ų��ڻ�ƽ��(12nλ)���м���(6mλ)��, ����������ͬһ��; ����Hԭ�ӵ�����, ��������aֵ(0.5273~0.5310nm)������Խ�С, ��cֵ(0.4075~0.4165nm)�нϴ������, ��ʵ����һ��; ����La��Ni������ý�һ��������
�ؼ���: LaNi5Hx; ����������; ��һ��ԭ��; ȫ����������ƽ�沨���� ��ͼ�����: TG139.7
���ױ�ʶ��: A
First-principle on hydrides LaNi5Hx for hydrogen storage process
ZHANG Chuan-yu1, GAO Tao1, QI Xin-hua1, CHEN Dong1,
ZHANG Yun-guang1, ZHU Zheng-he1, CHEN Bo2
(1. Institute of Atomic and Molecular Physics, Sichuan University,Chengdu 610065, China;
2. Institute of Nuclear Physics and Chemistry,Chinese Academy of Engineering Physics, Mianyang 621900, China)
Abstract: The density functional GGA method and the full-potential linearized augmented plane wave were used to optimize the cell and internal parameters of hydrides LaNi5Hx(x=3, 4, 5). The equilibrium structure, density of state and charge density were worked out. The results indicate that hydrogen atoms are distributed in the basal plane(12n site) and the middle plane (6m site) at end and tend to lie in the same side. With the increase of the number of hydrogen atom in the alloy, a value of crystal cell parameter (0.5273-0.5310nm)increases comparatively less whereas c value (0.4075-0.4165nm) increases comparatively more, which agrees exactly with the experiment; moreover the interaction between La atom and Ni atom bring down further.
Key words: LaNi5Hx; La-based hydrogen storage material; first principle; full-potential linearized augmented plane wave (FLAPW)
���������Ϊһ�ֶ���;����, �����������ǵ��ձ��ע, �о�������ϵ��۽ṹ�Լ������״̬�����˽�Ľ�������ϵ�����������Ҫ�����塣 LaNi5��AB5�ͻ������н�Ϊ����Ķ�Ԫ�������, ��ʵ��������϶��õ��˹㷺���о��� �ڴ���Ĺ�����, ������Ũ�ȵ�����, һ����Ϊ������ϴӹ�����(����)�����м����(��+����), ����⻯��(����)�� ���������о�[1-4]����, LaNi5-H2��ϵ�ڴ����м�����г��˹�������(����)����ȫ�⻯����(LaNi5H6)֮���һ���������⻯����LaNi5Hx(x��5), Ҳ����Ϊ�м��ࡣ ��Щ�⻯��������ͷ���ıؾ�����, ���������ǵ��о���ԱȽ��١� Ŀǰ����������LaNi5H3, Nomura��[1]������ƽ��ѹ�еĵ�������313Kʱ, LaNi5H2ϵͳ�д�������ƽ̨��, ��Ӧ�ķ���ʽ�ֱ�ΪLaNi5H��3��LaNi5H��6�� Akiba��[2]ͨ������ϵѹ������ֵ����ߵľ�ȷ����, ������һ���µĻ�������LaNi5H3 , X�����������֤��, ����������༰�⻯�������ͬ����������ṹ�� ����LaNi5H4, Matsumoto��[3]ͨ��X�����������, �ڴ��������, �����˽ӽ���LaNi5H4���ࡣ Lartigue��[4]����������ķ����о���LaNi5D5��һЩ���ʡ� ����, �����м������ϵ���⻯���ڲ�Hԭ�ӵ���״̬�ͽṹ����, Ŀǰ�����о������� ����Ӧ��Щ���, ���ɶ��Ż����ϵ�������������Լ������²��ϵ������������ġ�
�����������ܶȷ��������, ����ȫ����������ƽ�沨(FLAPW)����, �����о���LaNi5Hx(x=3, 4, 5)�ľ���ṹ�� ̬�ܶȺ͵����ܶȡ�
1 ����ģ���뷽��
1.1 ����ģ��
LaNi5��һ�־���CaCu5�;���ṹ��ϡ������Ͻ� ����Laռ1aλ(0, 0, 0), Niռ2cλ(13, 23), 0)��3gλ(12), 0,(12)), ����������ϵ, �ռ�ȺΪP6/mmm, �����ϵ�Ϳռ�Ⱥ���䡣
1.1.1 LaNi5H3��ģ��
һ��LaNi5�����й���37����϶, ����Westlake[5]����ľ������: ��ĸ�����������, ��ԭ����������λ���ǰ뾶r��0.04 nm���������϶�� Tatsumi��[6]����ó�: 12nλ���������, ���Ϊ6mλ�� Ono��[7]�ļ�����LaNi5H3�γ�ʱHԭ����Ҫռ�ݿ�����ƽ��(12nλ), �� LaNi5H3������ģ��1��ģ��2��
1.1.2 LaNi5H4, 5��ģ��
����LaNi5H3��LaNi5H7�ļ�����[8]�ȽϿ�֪, Hԭ�������Ų��������ϲ�ļ�϶, ��������ռ��12n��6mλ�� ��LaNi5H4��LaNi5H5�ֱ���������3��ģ�͡�
������̽�����������ģ�͵ľ�����������ԭ���ڲ����������Ż�����, �����м��������Ż������
1.2 ���㷽��
Ϊ�˼���LaNi5Hx(x=3, 4, 5)�ĵ��ӽṹ������, ���Ľ�����ȫ����ˮƽ�µ�ȫ����������ƽ�沨����(FLPW)�� �÷����Լ��㾧��ĵ��ӽṹ�dz���ȷ, ���Ǵ��ܶȷ�������Ϊ�����ĵ�һ��ԭ������, ���Ƚ��������ӷ��̻�Ϊ�����ӷ���(Kohn-Sham����), Ȼ��ͨ������������ƽ�沨�������Ե����ӷ��̽��м���, ͬʱҲ�����˹����ݶȽ�������(GGA)�������ӵĽ����ܱ�ʾ�ɵ����ܶȺ��ݶȵĺ���, ���ڸ����в�ͬ�ļ���, ���IJ��õ���Perdoew96�� ��������ƽ�沨��������ѡȡ�ͶԵ����ӷ�����Ǣ�����ʱ����ģ�͵Ľ����������ˡ�Muffin-Tin��ģ��[9]��
���ļ������WIEN2K����, �������㷽���Ѿ������ڸó����С�
2 ���������
�������еļ��㶼�����ܶȷ�������½��е�, �ڼ��㷽��Kohn-Shamʱ, ������Muffin-Tin ģ�ͶԲ�ͬ����ĵ���ʹ�ò�ͬ�IJ�����, ����La ��Niԭ�ӵ�RMT��Ϊ0.095nm, H ԭ��Ϊ0.037nm ; �ڽ����ܵļ�����ȡ-81.6eV ���ڲ���Ӻͼ۵��ӷֿ�, �����ӵĵ�����̬Ϊ: La5s25p65d16s2; Ni-3p63d84s2; H 1s�� �ڳ�ʼ��ʱ, ����Ԩ���в���k����ȡΪ500, �������������Ǣ�������������������ȿ���Ϊ: 1.36��10-3 eV�� ���ڼ����ж����ڲ���ӵļ������������۵��ܶȷ�������, ���ڼ����п����������ЧӦ��
2.1 �ṹ�Ż����
�ṹ�Ż�ʱ, ���ȱ���c/a����, �ı����; �������������, ���ı�c/a��ֵ, ���ּ���ֱ��η�������, �ڽṹ�Ż���ͬʱ������ţ����ѧ������ÿ��ԭ��ƽ��λ�ý����˳����Ż�, ֱ��ǰ�����ν���ļ���֮��С��1%��
�Ż����H��λ�ü���1~3, ���ںϽ����������, ģ������ԭ�ӵ�λ���ڼ�϶��Χ�ƶ�, �����ȶ��طֲ��ڻ�ƽ����м�����, ���Ҷ��ֲ��ھ�����ͬһ��, ����������������������˲�ļ�϶�����ɵ�, ������Tatsumi��[6]����LaNi5D6.7��ʵ����������˼��ּ���������⻯��ģ��, �õ���Ϊ�����˫�����ȶ��ṹ���Ǻϡ�
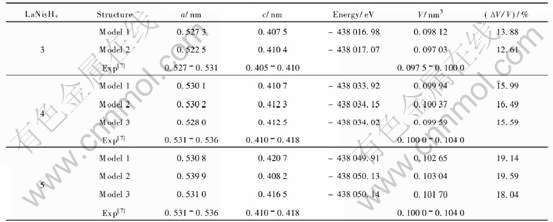
�ӱ�4�Ż�������Կ���, ����LaNi5H3, ģ��1��ģ��2����֮�����0.09eV, ������[6]��
��1 LaNi5H3�����е�Hԭ��ռλģ��
Table 1 Hydrogen location in unit cell of LaNi5H3
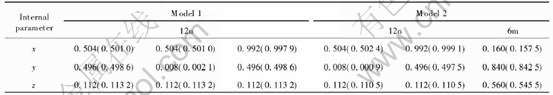
��2 LaNi5H4�����е�Hԭ��ռλģ��
Table 2 Hydrogen location in unit cell of LaNi5H4

��3 LaNi5H5�����е�Hԭ��ռλģ��
Table 3 Hydrogen location in unit cell of LaNi5H5
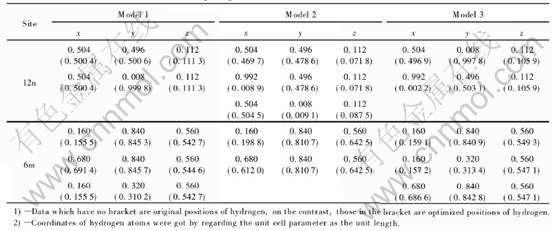
���һ��, ������ LaNi5[9]������;����������, ����ģ�͵�����;����������в�ͬ�̶ȵ�����, ��ģ��1��ʵ��ֵ���ϵĽϺ�, ����ģ��1Ϊ���������·����� ����LaNi5H4, 3��ģ����ʵ��ֵ��������, ��ģ��2���������, ����ģ��2Ϊ��������ķ����� ����LaNi5H5, �����������������ͺ������һ������, ������ģ��3���������, ����ģ��3Ϊ��������ķ�����
ͨ���Ա�1~4�����ɵ�, ����Hԭ�ӵ�����, ������һ������, ��������aֵ��0.5273����0.5310nm, �����С������, ��cֵ��0.4075����0.4165nm, �������ƱȽ�����, ��ʵ����һ��[6]; ���Ҿ������������, �������ž������������, �⻯�������ȶ�[10], ��˺Ͻ���ȶ�����Hԭ�ӵ����Ӷ����ӡ�
2.2 ̬�ܶȵķ���
̬�ܶȶ��ڷ��������е�ԭ�ӳɼ��Ͳ�����������Ҫ�����塣 ���ĸ�����LaNi5Hx(x=3, 4, 5)�ķ����ܼ��Լ�ͶӰ̬�ܶ�ͼ��
��4 LaNi5Hx(x=3, 4, 5)�ľ��������������Լ�����仯���Ż����
Table 4 Optimized results for unit cell parameters,energy and volume expansion of LaNi5Hx(x=3, 4, 5)
ͼ1��ʾΪLaNi5Hx(x=3, 4, 5)������̬�ܶ�ͼ�� �Է����ܼ�(EF)Ϊ0��, ����LaNi5H3��EF=-9.4342eV, LaNi5H4��EF=-9.3718eV, LaNi5H5��EF=-9.1949eV, ��LaNi5���[9] �����ܼ�������, ����������, �����һ������, ��Ҳ���������������ƽ̨ѹ���ߵ�ԭ��֮һ�� ����Hԭ�ӵ�����, �Ͻ������̬�ܶ���EF������; ����LaNi5H3��-5.77~-5.30eV֮���п���Ϊ0.47eV�Ľ���, LaNi5H4�Ľ���������-5.34~-4.66eV֮��, �������Ϊ0.68eV, LaNi5H5��-5.05~-4.33eV֮��Ҳ����һ����, �����Ϊ0.73 eV, �����Ŀ������, ���������c�����ͽϴ���ɵġ� ����������̬�ܶȵĿ����������Խ�С, �����������a�����ͽ�С��ɵġ�

ͼ1 LaNi5Hx(x=3, 4, 5)������̬�ܶ�
Fig.1 Total DOS of LaNi5Hx(x=3, 4, 5)
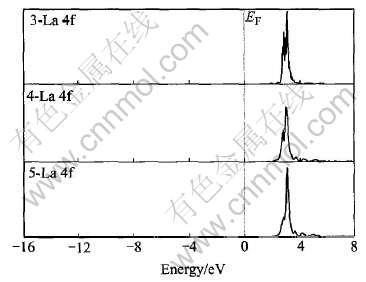
ͼ2��ʾΪLaNi5Hx(x=3, 4, 5)��La��4f�����̬�ܶ�ͼ�� ��̬�ܶ���Ҫ�����ڼ۴�3.0eV ����, ��ͼ1���, �۴���Ҫ����La��4f����ṩ�ġ� ͼ3��ʾΪLaNi5Hx(x=3, 4, 5)��Ni��3d�����̬�ܶ�ͼ�� ��ͼ1���, Ni��3d���ռ���˵����ľ���Ȩ��, ����EF����̬�ܶ���Ҫ����Niԭ���ṩ�� ͼ4��ʾΪLaNi5Hx(x=3, 4, 5)��H��s �����̬�ܶ�ͼ�� ����Hԭ�ӵ�����, ̬�ܶ���EF������, ����֮��Ľ�����խ��
ͼ2 LaNi5Hx(x=3, 4, 5)��La�IJ���̬�ܶ�
Fig.2 Partial DOS of La in LaNi5Hx(x=3, 4, 5)
ͨ��ͼ1~4���Եó�, La��̬�ܶ��������һ��ƽ��, ��Ni��H��̬�ܶȱ仯�����෴, �������һ���ƶ�, ��Ҳ�ܷ�ӳ��H��Ni�Ľ��ǿ��Ҫ����La�Ľ��ǿ�ȴ�, ����La��4f�����Ni��3d����ɼ�������, ���ֳɼ����������������������ɺϽ�������������ַۻ���ԭ��֮һ[11]�� �����ڽ�������̬�ܶȵ�չ����Ҫ����Ni��d�����H��s�� p����ṩ, ��Laԭ�ӵ�p���Ҳ�ṩ�������ĵ��ӡ� �ɼ�, ����Hԭ�ӵ�����Hԭ����Niԭ��֮�������γɹ��ۼ�[12], ��Laԭ����H ԭ���нϴ�ĵ縺�Բ��[13], �������֮���������ʾ������ǿ�ڹ����ԡ�
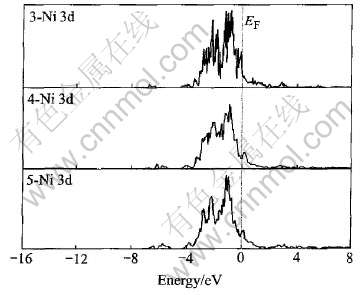
ͼ3 LaNi5Hx(x=3, 4, 5)��Ni�IJ���̬�ܶ�
Fig.3 Partial DOS of Ni in LaNi5Hx(x=3, 4, 5)
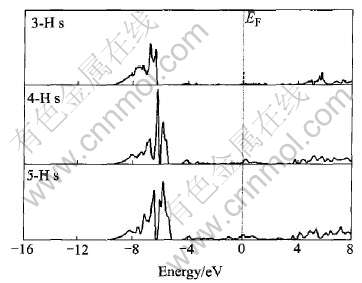
ͼ4 LaNi5Hx(x=3, 4, 5)��H�IJ���̬�ܶ�
Fig.4 Partial DOS of H in LaNi5Hx(x=3, 4, 5)
2.3 ����ܶȵķ���
����ܶȵĿռ�ֲ�, ���ڷ������ϵ�ԭ��֮��ɼ��Լ���������Ҳ����Ҫ�����á� LaNi5Hx(x=3, 4, 5)�Ż���ĵ����ܶ��ڿռ�ķֲ���ͼ5~7��ʾ��
��ͼ5(a)��ʾΪLaNi5H3����ĵ���ܶ�ͶӰͼ�� ���Կ���, Hԭ��λ�ڿ��������λ��, ��������Χ�нϸߵĵ���ܶȷֲ��� ����Hԭ�ӵĵ���ܶȷֲ���Niԭ�ӵĵ���ܶ������Ե��ص�, ����Laԭ�ӵĵ�ɷֲ�û���ص�������, ����Hԭ��������Χ��Niԭ�������γɹ��ۼ�, ����Laû��ֱ�ӵijɼ����á� ��ͼ5(b)���Կ���, ��Hԭ�Ӿ���Laԭ�ӽϽ�, ��������ܶ�ȴ����Niԭ�ӷ�����ɢ, ��Ҳ˵����H��Ni�Ľ��ǿ��Ҫ����La�Ľ��ǿ�ȴ�
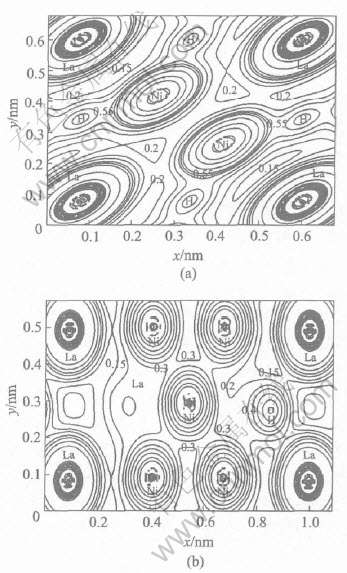
ͼ5 LaNi5H3��(0001)���(![]() )��ĵ���ܶ�
)��ĵ���ܶ�
Fig.5 Charge density in plane (0001) (a) and plane ![]() (b) of LaNi5H3(unit: 103 e/nm3)
(b) of LaNi5H3(unit: 103 e/nm3)
ͼ6(a)��ʾΪLaNi5H4�� (0001) ��ĵ���ܶ�ͶӰͼ�� ����Hֻ�Ƿdz���������, ������ʾ��Hԭ����Χ�ĵ���ܶ����������Щ, �����ڸ��������нϴ�ĵ���ܶȷֲ��� ��ͼ6(b)���Կ���, �����ڵ�λ����La�ĵ����ܶȽϵ͵ļ�϶λ��, ��Ҳ������H����ռ�ݰ뾶r��0.04 nm���������϶[5]��ԭ��

ͼ6 LaNi5H4��(0001)���(![]() )��ĵ���ܶ�
)��ĵ���ܶ�
Fig.6 Charge density in plane (0001) (a) and plane (![]() ) (b) of LaNi5H4(unit: 103 e/nm3)
) (b) of LaNi5H4(unit: 103 e/nm3)
��LaNi5H7���[8], Hԭ����Niԭ�ӵĵ�����Ȼ�������ԵĽ���, ����������γɹ��ۼ���ͼ7(a), ����Hԭ��λ�ڸ�����, ��������Χ�ĵ����ܶ����߽϶�, ������Χ�����ܶȺܴ� ͬʱ����������ͽϴ�, ʹLaԭ����Niԭ��֮�������ý�һ������(ͼ7(b))��
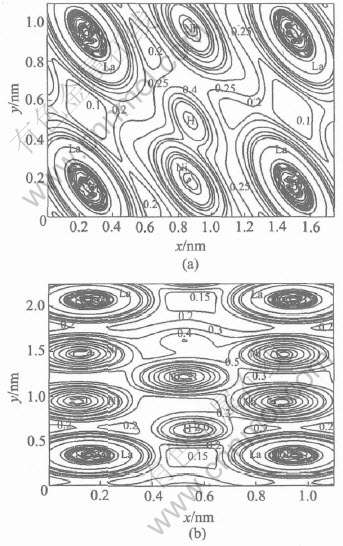
ͼ7 LaNi5H5�� (010) ���((110))��ĵ���ܶ�
Fig.7 Charge density in plane (![]() ) (a) and plane (
) (a) and plane (![]() ) (b) of LaNi5H5(unit: 103 e/nm3)
) (b) of LaNi5H5(unit: 103 e/nm3)
��ͼ5~7�ĵ����ܶȷ����ɵ�, ���źϽ���H�ĺ���������, ��ͼ���Ͽɿ���Hԭ����Niԭ��֮��ijɼ����, ��Laԭ����Ϊ�⻯����γ�Ԫ��[14, 15], ��Ȼ��Hԭ��֮����нϴ������, ��������֮��ɼ�������
3 ����
1) �ӵ�һ��ԭ������, �״ζԴ����м�����д��ڵ��⻯��LaNi5Hx(x=3, 4, 5)�ľ���ṹ�Լ��ȶ��Խ����˽�ģ�о�, ��������Ӧ�ľ���ṹ�� ̬�ܶ��Լ�����ܶȡ�
2) ��LaNi5Hx(x=3, 4, 5)�Ľṹ�Ż�����ó�, ��ԭ���ȶ��ķֲ��ڻ�ƽ����м�����, �������ھ�����ͬһ�ࡣ ����Hԭ�ӵ�����, ��������a��������Խ�С, ��cֵ���������ƱȽ�����, ���ҽ����⻯���ȶ�����ǿ��
3) ��LaNi5Hx(x=3, 4, 5)��̬�ܶȺ͵����ܶȷ�������, H����Ni�����γɹ��ۼ�, ����La�ɼ����������; Laԭ����Niԭ��֮������������Hԭ�ӵ����Ӷ�����������
REFERENCES
[1]Nomura K, Uruno H, Shinozuka H, et al. Effects of lattice strain on the hysteresis of pressure-composition isotherms for the LaNi5-H2 system[J]. J Less-Common Met, 1985, 107: 221-230.
[2]Akiba E, Nomura K, Ono S. A new hydride phase of LaNi5H3[J]. J Less-Common Met, 1987, 129: 159-164.
[3]Matsumoto T, Matsushita A. A new intermediate hydride in the LaNi5-H2 system studied by in situ X-ray diffractometry[J]. J Less-Common Met, 1986, 123: 135-144.
[4]Lartigue C, Percheron A, Achard J C. Hydrogen ordering in the ��-LaNi5Dx>5 phases: A neutron diffraction study[J]. J Less-Common Met, 1985, 113: 127-148.
[5]Westlake D G. A geometric model for the stoichiometry and interstitial site occuping in hydrides of LaNi5, LaNi4Al, LaNi4Mn[J]. J Less-Common Met, 1983, 91: 275-292.
[6]Tatsumi K, Tanaka I, Inui H, et al. Atomic structures and energetics of LaNi5-H solid solution and hydrides[J]. Phys ReV B, 2001, 64: 184105.
[7]Ono S, Nomura K, Akiba K. Phase transformations of the LaNi5-H2 system[J]. J Less-common Met, 1985, 113: 113-117.
[8]Hector L G Jr, Herbst J F, Capehart T W. Electronic structure calculations for LaNi5 and LaNi5H7: energetics and elastic properties[J]. J Alloys Comp, 2003, 353: 74-85.
[9]���»�, ����, ��. LaNi5�����������ṹ��ȫ���Ӽ���[J]. ԭ�ӷ�������ѧ��, 2004, 21: 366-372.
QI Xin-hua, GAO Tao, et al. Full-electronic calculations on the equilibrium structure and energy of LaNi5 crystal[J]. J At Mol Phys, 2004, 21: 366-372.
[10]Mendelsohn M H, Gruen D M, Dwight A E. The effect of alumimum additions on the structural and hydrogen absorption properties of AB5 alloys with particular reference to the LaNi5-xAlx ternary alloy system[J]. J Less-Common Met, 1979, 63: 193-207.
[11]Τ��¥, ����, ����, ��. LaNi5���Ͻ�缫����������ӽṹ�������[J]. �й���ɫ����ѧ��, 2002, 12: 501-504.
WEI Wen-lou, GUO Jin, DENG Wen, et al. Correlation between electronic structure of LaNi5 base alloys and hydrogen absorption properties[J]. The Chinese Journal of Nonferrous Metals, 2002, 12: 501-504.
[12]����, Τ��¥, ����Ԫ, ��. LaNi5���ӽṹ��ɼ�����[J]. ����ѧ��, 2003, 39: 10-12.
GUO Jin, WEI Wen-lou, MA Shu-yuan, et al. Electronic structure and bond character of LaNi5[J]. Acta Metall Sin, 2003, 39: 10-12.
[13]Nakamura H, Nguyen-Manh D, et al. Electronic structure and energetics of LaNi5, ��-La2Ni10H and ��-La2Ni10H14[J]. J Alloys Comp, 1998, 281: 81-91.
[14]van Vucht J H N, Kuipers F A, et al. Reversible room-temperature absorption by intermetallic compounds[J]. Philips Res Repts, 1970, 25: 133-140.
[15]Yukawa K, Nakatsuka K, Morinaga M. Design of hydrogen storage alloys in view chemical bond between atoms[J]. Solar Energy Materials & Solar Cells, 2000, 62: 75-80.
������Ŀ: ������Ȼ��ѧ-�й����������о������ϻ���������Ŀ(10276027)
�ո�����: 2005-09-07; ������: 2005-11-20
ͨѶ����: ����, ������; �绰: 028-85405234; E-mail: gthhl@sina.com
