
���±�ţ�1004-0609(2010)12-2357-09
п�����Pb/Pb-MnO2���ϵ���������Ʊ�������
�� Ԩ1��������1���ߺ㷢2��������1��������1���� ��1����ҵ��1
(1. ���ϴ�ѧ ұ���ѧ�빤��ѧԺ����ɳ 410083��2. ����ԥ��пҵ����˾����Դ 464530)
ժ Ҫ��
���ø��ϵ�Ƽ����Ʊ�Pb/Pb-MnO2���ϵ���������о�MnO2���������������缫���á�ʩ��ʱ������Ӽ��ȶԶƲ�����������Ӱ�죬��������ʵ��ȷ�������Ʊ�����������������ѭ�������ͺ����������ֶζԸ�������������������ܽ������ۡ����������ͨ�������Ż������ڷ������ζ�Ǧ��ϵ��ʵ��Pb������Pb-MnO2���϶ƣ����MnO2������Ϊ1%~10%(��������)�ĸ��϶Ʋ㣻ģ�ҵп��������У�����ʼ�θ���������λ���Ǧ��������Լ300 mV������24 h�������ȶ���λ�Ա�Ǧ������100 mV���빤ҵPb-Ag(0.6%����������)�������ȶ���λ�൱��
�ؼ��ʣ�
п��������ϵ����Pb/Pb-MnO2����������������ϵ�����������
��ͼ����ţ�O646���� ���ױ�־�룺A
Preparation and properties of Pb/Pb-MnO2 composite anode for
zinc electrowinning
LI Yuan1, JIANG Liang-xing1, NI Heng-fa2, L? Xiao-jun1, LAI Yan-qing1, LI Jie1, LIU Ye-xiang1
(1. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China;
2. Henan Yuguang Zinc Industry Co., Ltd., Jiyuan 464530, China)
Abstract: A new type of Pb/Pb-MnO2 composite anode was prepared by composite electrodeposition, the effects of particle size and crystal of MnO2, electrodes configuration and electrodeposition time on the particles content in co-deposit were studied, and the optimum conditions of anode preparation were ascertained by orthogonal experiments. The electrocatalytic activity for oxygen evolution reaction (OER) of the composite anode was evaluated by cyclic voltammetry (CV) and galvanostatic polarization method. The results indicate that Pb/Pb-MnO2 composite deposit containing 1%~10% (mass fraction, %) MnO2 particles can be obtained in the fluoborate solution of lead plating. In the stimulant evaluation of industrial electrowinning, the composite anode prepared under the optimal conditions is about 300 mV, more active than the pure lead anode at the initial stage of electrolysis, and the stable potential after 24 h is 100 mV lower than that of pure lead anode, which is equal to that of Pb-Ag (0.6%) anode.
Key words: zinc electrowinning; composite electrodeposition; Pb/Pb-MnO2 anode; fluoborate system; oxygen evolution electrocatalysis
����������ʪ����п��ҵ�ĵ������һֱ����Pb-Ag(0.6%~1.0%����������)�Ͻ�����[1-2]������Ȼ������п����Ļ���Ҫ���Դ�������㣬����������λ��(860 mV)��Ǧ�ĸ�ʴ�������Ⱦ����п��������������(������Cl-����ʱ)�����⣬Ǧ�ܶȴ�еǿ�ȵͣ�������䣬���������������·�����͵�Ч��������ϲ��㣬�����о��˶�������������ͼ���ƻ������ͳPb-Ag��������Ҫ�����ڶ�Ԫ�Ͻ� ��[3-8]�ͱ���Ϳ������Խ���������[9-14]�������棬����Щ�����ʹ�����������̸��ӣ��ɱ���ߣ�����
��������ײ���(��Ti)�������ۻ���Ϳ�����ױ��ƻ���ʹ��������������������ģ��Ӧ���ڹ�ҵ�����С�����ѧ��[15-17]�����ϵ�Ƽ���Ӧ����ʪ����������������Ŀ����У��Ʊ���Pb/Pb-Co3O4��Pb-Ca/Pb-CoTiO3��Al/Pb-Y(Y=WC��ZrO2��CeO2��Ag)�ȸ���Ϳ�����������ִ�����������һ���̶��Ͻ���������λ����С��ʴ���ʣ�ʹ�������ϳ�����ˣ�Ѱ����Ч���������������ø��ϵ�Ƽ����Ʊ����ߵ�����Ը���������һ����������DZ�����о�����
MnO2����õ���������֮һ��������Ը��ҳɱ�������SCHMACHTEL��TOIMINEH[18]������Ϳ����(Cold spray technique)�Ʊ���Pb/Pb-MnO2�����������������ڵ�����������λ��Pb-Ag(0.6%)�����ĵ�250 mV�����ֳ����������������ܡ�������Ϳ���̲������ӣ��Ʊ��ɱ��ߣ���Ʒ�п���֮�俿���Ա���ճ����һ��δ�γɾ�����ʣ�����ȱ�ݶ࣬������ʴ�Բ�����ȣ����ϵ�Ƽ���[19]���ռ������������ڿ��ƣ�����Ʒ�Ʊ������в�������ԭ������(�羧�ͺ���ò��)�ĸı䣬�γɾ�����ʾ��нϺõ���ʴ�ԣ�����������Ϳ�IJ��㡣��ˣ��������߲��ø��ϵ�Ƽ����Ʊ��ߴ����Ե�Pb/Pb-MnO2���������������临�ϵ�ƹ��պͶƲ�ĵ绯ѧ���ܽ����о���
1 ʵ��
���ϵ����ϵ��������������Ϊ20 mm�� 20 mm��3 mm�Ĵ�Ǧ�飬���þ�Ե���ܷ⣬ֻ����20 mm��20 mm�Ĺ��������ʵ����250 mL�ձ��� ���У�����DF-101S����ʽ���´�������������ԴΪ��γGPS1830Dֱ����ѹ��Դ�������Լ���Ϊ��������MnO2���ȴ���99.5%(���ϻ�ͨ�Ƽ�����˾)����Һ��ȥ����ˮ���ơ�
���ϵ��Һ�Ļ���������£�Pb(BF4)2 120~150 g/L��HBF4 30~40 g/L��H3BO3 12~15 g/L��һ������MnO2�����������Ӽ���
���ϵ�Ƶľ��岽�����¡�
1) ��ǰ�������� �����Ƶ缫������220#��400#ɰֽ�Լ�����W28(01)ɰֽ��ĥ��������Ȼ����ȥ����ˮ��ϴ���� ��NaOH��Na2CO3��Na3PO4��Na2SiO3����Һ�н���10~15 min������ȥ����ˮ��ϴ���� ��30%������Һ����5~10 min������ȥ����ˮ��ϴ��
2) ���ϵ�ƣ��� �����⡣��װʵ��װ�ã���������������2 A/dm2���������缫3~5 min���� Ԥ�ƴ�Ǧ�����ӵ�·����4 A/dm2�����������ϵ��30 min���� ���ϵ�ơ����������������������裬��Ԥ���������ϵ�ơ�
3) �ƺ������� �����϶Ʋ��ڼ�ˮ�г�ϴ3~5 min���� ����ˮ�ƾ��н���5~10 min������紵�ɺ档
����JSM-6360��ɨ��羵(SEM)�۲츴�϶Ʋ�ı�������ò��GENESIS60S��X������������(EDX)�ⶨ��Ԫ����ɣ�����ʽ(1)����Ʋ���MnO2��������������
![]() (1)
(1)
ʽ�У�MPb��MMn��MO�ֱ�ΪPb��Mn��O�����ԭ��������n(Pb)/n(Mn)ΪEDX�ⶨ��Pb��MnĦ���ȡ�
�������缫��ϵ�Դ�Pb��������ҵPb-Ag(0.6%)������Pb/Pb-MnO2�����ֱ����ѭ�������ͺ����������ԣ��ԱȽ��������ܡ������й����缫Ϊ�������������������缫���ò�Ƭ���αȵ缫���ñ��ʹ��缫(SCE)�����õ��Һ���Ϊ250 g/L ZnSO4+160 g/L H2SO4�����200 mL��ѭ������������Parstat2273�绯ѧ�ۺϲ������Ͻ��У�ɨ������0~2.2 V��ɨ������10 mV/s���������������������ܶ�Ϊ5 A/dm2������ʱ��Ϊ24 h���¶Ȳ���DK-S24�͵��Ⱥ���ˮԡ��������(35.0��0.5) �档
2 ���������
2.1.1 �缫������ʽ��Ӱ��
���ϵ���е缫�Ĺ�����ʽͨ�������֣�ƽ�õ缫(Horizontal electrode)�����õ缫(Vertical electrode)��ZAHAVI��HAZAN[20]��Ni-���ʯ���ϵ�Ƶ��о���ָ������ƽ�õ缫���������������Դ������õ缫����ʵ����Ҳ�������ֵ缫���ý������о�����Ӧ�Ʋ������ò��ͼ1��ʾ(��ɫ�Ķȣ�MnO2�������Ұ�ɫ�Ķȣ�Pb����)��
ͼ1��ʾΪ�缫���öԶƲ������ò����������Ӱ�졣ͼ1(a)��MnO2��(����Ϊ7.89%����������)��ֲ��ھ���֮��Ĺ����У���ͼ1(b)��MnO2

ͼ1 �缫���öԶƲ������ò����������Ӱ��
Fig.1 Influence of electrode configuration on surface morphologies and particle contents of deposits (Other experiment condition: ��-MnO2 (2-7 ��m), particle suspension 80 g/L, current density 4 A/dm2, temperature 35 ��): (a) Horizontal electrode (experiment agitation rate 550 r/min); (b) Vertical electrode (experiment agitation rate 400 r/min)
��(����Ϊ4.83%����������)�ȽϷ�ɢ������Ҫ������2~7 ��m�����ڸ��϶��������ϴ������룬�������õ�Ӱ�첻�ɺ��ԡ��о�����[21]��Һ������ƽ�����ʱ���������ʮ������������߽�㣬�������Χ����������(��)��ȱ�������(��0)��ͣ��������еĴ��ʱ����⣬�ڵ缫���渽��������һ�㼸ʮ�����˫��㣬��������Ч��������Ҫ��Ӱ��[22]����ͼ2��ʾ�����ڶ�Һ����Ҫ������(Fg)������(Fp)����������(Fw)�����ã���˫����⣬�糡�����������Ժ��Բ��ơ��缫ƽ��ʱ(ͼ2(a))�����ڱ߽�����������ٽ��ͣ������ܳ������С�����������������³������缫�ϡ�ͬʱ��ƽ�õ缫����İ���ƽҲ�����ڻ�е��������ʹ�������Ƕ��Ʋ��У���֮���缫����ʱ(ͼ2(b))���߽���������ʱ���������ʹ��Ѹ�ٳ���������˫�����������٣�����ʱ�缫�����ֲ����ڻ�е��������ˣ��Ʋ��е���Ƕ�������١����⣬��ʵ�ʲ����У��缫����ʱ�������ʺ͵缫����ȶ���Ƕ���Ӱ���Ϊ���ӣ�������������ơ� ��ˣ��������߲���ƽ�õ缫���и��ϵ�ơ�
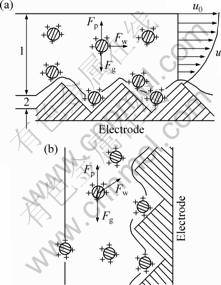
ͼ2 ��ͬ�����µ缫���渽����������״̬
Fig.2 Stress state of particles near electrode surface in different electrode configurations (1��Boundary layer; 2��Electrical double layer): (a) Horizontal electrode; (b) Vertical electrode
2.1.2 MnO2�����ͺ�������Ӱ��
MnO2�dz��õĵ�ز��ϣ�Ҳ����Ч������������������ж��־��ͣ�����Ҫ���Ǧ����¡��úͦ��͡��о�����[23]����ЩMnO2���ձ�ṹ��[MnO6]�����干������ö����γɱ仯��˵ĸ������磬�������ɸ��ֲ�ͬ�������Ӻ���λ�����̵��������ж��ֶ�������ɺ;���ṹ�����ʹ����Զ��ԣ���-MnO2�д��ڴ�����Mn3+�Ϳ�Ѩ��ʹ���䱾��ͱ��涼���ڴ����ġ�����۵��ӡ�����;������������൱�õĴ�����[24]����ʵ���Գ��õĦºͦ�����MnO2(����2~7��m)Ϊ��ɢ���������о����۲츴�϶Ʋ������ò(��ͼ3)���ⶨ���������������������߹�����������
ͼ3��ʾΪ�ֱ��Ԧ�-MnO2�ͦ�-MnO2Ϊ������ʱ����Ʋ�ı�����ò�����Ʋ��������ò������-MnO2(����Ϊ6.86%����������)�ڶƲ��е�Ƕ����ԶԶ���ڦ�-MnO2(����Ϊ1.69%����������)������Ҫ���������Ǿ���ṹ�ͱ���״̬�IJ��졣CELIS��[25]�岽��������ָ���������ڶ�Һ�е��������������ڱ����γ��ܼ���Ĥ������Ǩ�Ƶ��缫���淢������������MnO2���ԣ����������ͽ��������ںܴ�̶���ȡ���������ġ�OH�������ԡ�OH���е�H
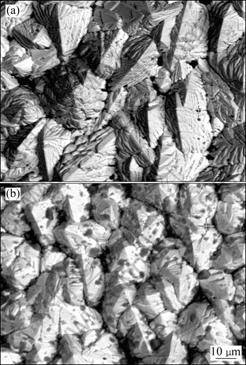
ͼ3 �ֱ��Ԧ�-MnO2�ͦ�-MnO2Ϊ��ɢ��ʱ����Ʋ�ı�����ò
Fig.3 Surface morphologies of deposits using ��-MnO2 and ��-MnO2 as disperse phase (Experiment condition: particle suspension 80 g/L, current density 4 A/dm2, temperature 35 ��, agitation rate 550 r/min, deposition time 60 min): (a) ��-MnO2; (b) ��-MnO2
�ɱ�����������(��Ni2+��Zn2+��Co2+��)ȡ����ʹ��������[26]����Թ������������ġ�����[23]ָ������-MnO2�DZ��Ľ��ʯ�ͽṹ�����������ԭ����ȣ��ᾧ�����ã����������ڱ���ͱ����������OH������������Բ�ڸ��ϵ��ʱ����Ƕ��Ʋ��У���֮����-MnO2�ľ���ṹ�ʽϴ��[1��2]�������ұ�����ڴ����ġ�OH�����ڶ�Һ�оͿ�������Pb2+��H+�������Ӷ������磬���䵼���������ڦ�-MnO2�ģ���ˣ���-MnO2�������������������淢����������
���⣬��ʵ���ڿ��Ƶ����ܶ�(4 A/dm2)���¶�(35 ��)��ʩ��ʱ��(60 min)��������ͬ��ǰ����(�������ʵ��������պò��ѻ����缫����)���о��˦�-MnO2����(20~40 ��m��2~7 ��m��40 nm)�Ը��϶Ʋ�����������Ӱ�졣���������ֻ��2~7 ��m�Ħ�-MnO2���ܴ�����Ƕ�뵽�Ʋ���(����Ϊ5.86%����������)��40 nm�Ĵ�֮(����Ϊ1.49%����������)����20~40 ��m�����������ܽ���Ʋ�(����Ϊ0.47%����������)�����ܵ�ԭ�����£�����MnO2�����ܸܺߣ�����Һ�м����žۣ���Һ����������һ�㲻Ӧ����10 g/L��������õĶƲ��������ͱȽ�С���������ڶ�Һ���ȶ��Ժã��ž�С�������ںܸߵ�����Ũ��(80~125 g/L)�½��и��϶ƣ��Ӷ�����������ߵĶƲ㣻������������20~40 ��mʱ����Һ�����ij�ˢ�������ԣ����Ѿ����ѱ��Ʋ�������γ���Ч�İ��������Ƕ�������١���Ȼ���������ڶ�Һ�е�����״̬��ͨ�����ӱ�����Լ��ͼ�ǿ����ȴ�ʩ�����ƣ��Ӷ�����������ߡ������Ժõĸ��϶Ʋ㣬�б�Ҫ���������̽�����ڱ��о��У���������˵�������������������Ϊ2~7 ��m�Ħ�-MnO2��
2.1.3 ʩ��ʱ���Ӱ��
��Pb/Pb-MnO2���������Ϸ���������Ӧʱ��ʵ����ֻ�б��㸽���ķ�ɢ����������ã��Ʋ��ڲ������������١�ͬʱ��MnO2����һ�ֵ����ԱȽϲ�IJ��ϣ��ڶƲ��л����ӵ缫���裬�������������λ�����ԣ��������������Ч��ʵ������MnO2������Ժ͵����Ե��ۺϽ���������о�����[22]���ڸ��ϵ�Ƶij�ʼ�Σ��Ʋ����������ر�ߣ�����ʩ��ʱ������ӣ����������٣��ڱ��о���Ҳ�ó������ƵĹ��ɣ������ͼ4��5��ʾ������Ҫ����Ϊ����ʩ��ʱ������ӣ��绯ѧ��ƽ����ʹ�缫����ֲڶ����ͣ������������٣���Һ�и�Ũ�ȵ�MnO2�����������žۣ�ʹ����Ʋ�������١���ˣ���ͨ����������ʩ��ʱ�������ƶƲ�ĺ�ȣ�ʹ����MnO2�����㹻�ߣ��ҵ缫����Ҳ�Ƚϵ͡�
��ͼ5�ɼ�������ʩ��ʱ�����ӣ��Ʋ���ƽ��������Ƕ����������������С����������֤һ�¡����⣬��ʩ��ʱ��Ϊ60~90 minʱ(��ͼ4)�����߳���ƽ̨��������Ǿ���һ��ʱ����ݺ����ڶ�Һ�е�ʪ���Ա�ã��ܸ��õ������ڵ缫���棬����ٽ��˹��������绯ѧ���Է��֣�ʩ��60~90 minʱ�������������������Ч�����ȽϺã���˱��о��и��ϵ��ʱ��ѡ��Ϊ60 min��
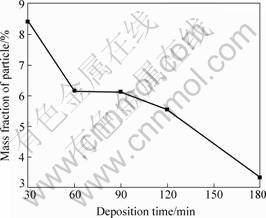
ͼ4 ʩ��ʱ��Ը��϶Ʋ�����������Ӱ��
Fig.4 Influence of deposition time on particle contents in deposits

ͼ5 ��ͬʩ��ʱ��ʱ���϶Ʋ�ı�����ò
Fig.5 Surface morphologies of deposits at different deposition time: (a) 30 min; (b) 90 min; (c) 180 min
2.1.4 ����ʵ��ȷ�����Ż�����
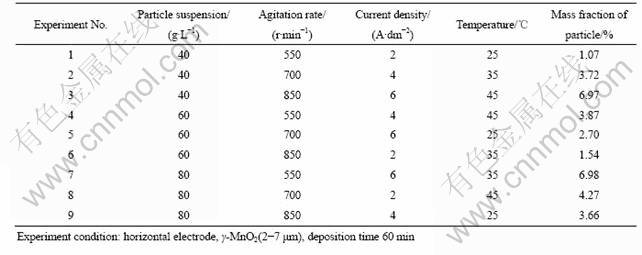
�������Ͻ��ۣ���ʵ����ϵ�Ƶ�������������(��Һ�����������������ܶȡ��������ʺ��¶ȵ�)�ԶƲ�����������Ӱ�죬��չ����ʵ�飬�Ի�ø��������Ʊ�����������������ʵ�����������1���С�
ʵ���о����֣��������ضԸ��϶Ʋ�������������һ����Ӱ�죬��Ӱ��̶��ɴ�С����Ϊ�����ܶȡ��¶ȡ���Ũ�Ⱥͽ������ʡ�����ǰ������ֵ�������������ֱ������Ե����ӣ�ԭ��������£����ŵ����ܶȵ����ӣ������������������������Ҳ��֮��ǿ�������¶ȵ����ߣ���Һ�Ƚ��ͣ��������ٶ�Ҳ��֮���ӣ�������������ʹ��������Ӵ����缫���棬��Щ���Ƕ��������������ġ���Զ��ԣ��������ʶԶƲ�����������Ӱ���С�����������Ϊ�ڵ缫��������߽���ڣ����������Ѿ�����ʹ�㹻������������缫���淢��������������ʵ��ó����Ʊ������������϶Ʋ����������Ϊ����Һ����������80 g/L������550~700 r/min(����������С��550 r/minʱMnO2����ѻ��ڵ缫������ò����Ʋ�)�������ܶ�6 A/dm2���¶�45 �档
2.1.5 ���Ӽ��ԶƲ�����������Ӱ��
Pb2+���������λ�ܵͣ��缫��Ӧ�ٶȿ죬�ڶ�Һ�м����γɴֲڡ���֦״����״�ij�����������һ�����Ӽ����軯��������̣��Եõ�ƽ�������ܡ���ɢ�ԺõĶƲ㡣���⣬�ڸ��ϵ���У������Һ��
��1 ���ϵ������������Ƽ���Ӧʵ����
Table 1 Orthogonal experiment design of composite electrodeposition and corresponding results
���������ı�����Լ����ı�����ʪ���Ժͱ���ɵ�״̬��ʹ����õ�Ƕ��Ʋ���(�������ߣ��ֲ����ȣ�����ʽ�Ϻ�)���������߾������������ӱ�����Լ�A�����Ӽ��Ը��϶Ʋ������������������ò��Ӱ��������о�����ͼ6��ʾ�����������������ӣ��Ʋ������������ֳ���������С�����ƣ���0.5g/Lʱ��ߣ�ͬʱ�����Һ�����������ӱ�����Լ�A������(0.5 g/L)���ʹ�ã���ʹ�Ʋ�����������һ������(������Լ�A�ļ�������С��0.5 g/L��������������0.5 g/Lʱ���Ʋ���������������С)��
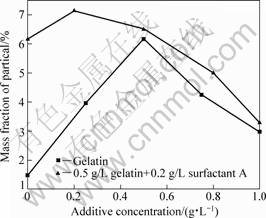
ͼ6 �����������ӱ�����Լ�A�ԶƲ����������� Ӱ��
Fig.6 Influence of gelatin and cationic surfactant A on particle content in deposit
������һ���ɶ��ְ�������ɵĸ߷��ӽ�ԭ���ף��������Ժͼ��ԡ�һ���棬���������������͵缫���棬��ǿ�����Һ���缫֮�����ʪ�ԣ����ڵ�����������軯�绯ѧ��Ӧ���õ��ᾧϸ�£���ɢ�ԺõĶƲ㣻��һ���棬�����Խ����У����������������Ӷ��γ�NH3+��R��COOH���൱��һ���������ͱ�����Լ���������������ʹ������磬�ٽ�����������Һ�в�������ʱ�õ��ĶƲ�ֲڣ�MnO2�������ܵͣ����京������0.5 g/Lʱ���õ��ĶƲ�����Ƚ�ƽ�����������ﵽ���ֵ�����������������ߣ��Ʋ��������������½���������Ƕ�Һճ������MnO2���ʱ�����缫�����������С���£���ͼ7(a)��(b)��(c)��ʾ��
�����������ӱ�����Լ�A���ʹ�õõ��ĸ��϶Ʋ����ƽ�������ܣ�������Ҳ�������ߡ�������Ϊ�����ӱ�����Լ���������Һ���Դ�������л������Ŵ��ڣ�������������MnO2������ʹ������磬һ����ٽ����ĵ�ӾǨ�ƺ��ڵ缫�������Ч�����������ڹ���������һ��������������ʽ�������ʪ���ã�ʹ�����Ա�ã���ͼ7(d)��ʾ��
���⣬�����л����Ӽ���Ϊ�����Խϲ�����ʣ������ڶƲ��л��������ӵ缫���裬ʹ������λ���ߡ�

ͼ7 ���Ӽ��ԶƲ������ò��Ӱ��
Fig.7 Influence of additives on surface morphologies of deposits: (a) 0 g/L gelatin; (b) 0.5 g/L gelatin; (c) 1.0 g/L gelatin; (d) 0.5 g/L gelatin+0.2 g/L surfactant A
��ˣ�ֻ�п���ǡ���ļ����������ܻ�õ��������õĸ��ϵ缫��
2.2.1 ѭ����������
ͼ8��ʾΪPb��Pb-Ag(0.6%)��Pb/Pb-MnO2�������ʵ缫������ǰ��ZnSO4+H2SO4��Һ�в�õ�ѭ���������ߡ���ͼ7�ɼ���Pb/Pb-MnO2��Pb��Pb-Ag�����ĵ绯ѧ��Ϊ������һ�£�������Ҳ��Ϊ���ԡ�
��ͼ8�ɼ�������ɨ��ʱ�����ȣ�����Pb��������H2SO4��ϵ���γ��˽�Ϊ���ܵ�PbSO4�ۻ�Ĥ����0~1.2 V�ĵ�ѹ������3�������ĵ����ܶȻ���Ϊ0��֮��Pb��Pb-Ag�������ȷֱ���1.85 V��1.76 V����������������(Anodic peak)��������λ���Ʋ�[27]���÷��Ӧ��ӦPbO����-PbO2����Σ�3�������ֱ���1.95~2.2 V��1.8~2.2 V��1.8~2.2 V�����������֦(Anodic branch)������PbSO4����-PbO2��H2O��O2������Ӧ���ӵĽ��[28]��Pb-Ag��Pb/Pb-MnO2������Ӧ�÷����Ը��ƣ�˵������ͬ�����ܶ��£����Ǿ�����Ч����������λ������Pb/Pb-MnO2����Ч����á����⣬Pb/Pb-MnO2��������û�� PbO����-PbO2�����壬���Ʋ�Pb��Pb-Ag�����ڼ�������������������ɦ�-PbO2�����������������������ɵ����ԺõĦ�-PbO2���������������Խϸߵ�ԭ��֮һ������ɨ��ʱ��������֦��࣬Pb/Pb-MnO2��Pb-Ag�������ȷֱ���1.44 V��1.48 V����һ�������壬������ǵ��Һ�е�SO42-ͨ���缫�����γɵ�PbO2�����뵽Pb�����Ϸ�����ӦPb��PbO2��PbSO4[28]����Pb����ȴ�۲ⲻ���˷壬���ܵ�ԭ�����£���Ǧ�������淴Ӧ���Եͣ��绯ѧ��Ӧ���������Ϲ۲ⲻ���÷�Ӧ�壻Ag�ļ����ṩ�˴����ķ�Ӧ���Ե㣬�ںܴ�̶��ϴٽ��˸÷�Ӧ�ķ����������������ӣ��������������ָ÷�Ӧ������ΪMnO2�ļ���ʹ�Ʋ��ôֲڣ������������ӣ��绯ѧ��Ӧ�����ӡ���Σ�3��������1.35 V���Ҷ����Լ�PbO2��PbSO4�Ļ�ԭ��(Cathodic peak)��Pb/Pb-MnO2��Pb�����Ļ�ԭ����������Pb-Ag�����Ļ�ԭ��߽ϴ�˵��Ag�ļ���ʹ������������(1.7~2.2 V)�����˽϶�����PbO2����Ҳ�����������Խϸߵ�ԭ��֮һ��ͬʱҲ����ӡ֤Pb/Pb-MnO2�ϸߵ�����������Դ��MnO2�Ĵ����á�����ָ�����ǣ�ͼ8�й۲ⲻ��MnO2����ط�Ӧ�壬��˵�����ڼ������������������ᷢ����ѧ�仯��ֻ��������Ӧ�����á�
2.2.2 ������������
��ҵп������ں㶨�����½��еģ�������λ�Ǻ����ù����ܺĵ�һ����Ҫָ�ꡣͼ9��ʾΪPb��Pb-Ag(0.6%)�������������Ƶ�Pb/Pb-MnO2��������24 h����������������λ���ߡ���ͼ9���Կ������ڵ���ʼ�Σ�Pb/Pb-MnO2������������λѸ�ٽ���(���Pb�����ɽ���Լ300 mV)������������Ժ���Pb��ҵPb-Ag(0.6%)����������ѭ���������߲ⶨ�Ľ��һ�£�������λ��һ���̶ȵĻ���������ȶ���λ����Pb����100 mV����Pb-Ag(0.6%)�����൱��
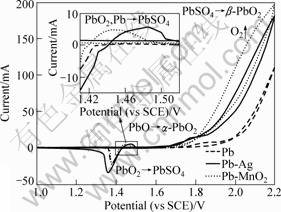
ͼ8 Pb��Pb-Ag��Pb/Pb-MnO2������ѭ����������
Fig.8 Cyclic voltammograms of Pb, Pb-Ag and Pb/Pb-MnO2 anodes
�Ժ���������ʼ�ε�������λ���߽��зŴ���Pb/Pb-MnO2�����ļ���������һ���ԵĹ��Σ���ɹ�����Pb�缫��H2SO4��ϵ�е��ʱ������״̬�����ı仯�������������������һ�����ɣ����PbO2Ĥ��һ���棬�������Ĥ���ܸ��Dz���MnO2
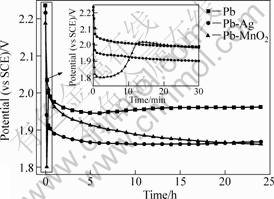
ͼ9 Pb��Pb-Ag��Pb/Pb-MnO2������24 h����������������λ����
Fig.9 Galvanostatic polarization curves of Pb, Pb-Ag and Pb/Pb-MnO2 anodes for 24 h
����ʹ�䲻������Һ�Ӵ���������绯ѧ��Ӧ����һ���棬����Ĥ������ʹ��Ƕ�ڵ缫�����MnO2������ɶ������������������Һ���������ij�ˢ���ø��Ӿ�����һ���̡����ԣ����ڱ���������ļ��٣�Pb/Pb-MnO2�����Ĵ����Ի���һ��ʱ����������ͼ10��ʾΪPb/Pb-MnO2������������12 hǰ��ı�������ò��EDX���Է�������MnO2�����ӳ�ʼ��6.24%���͵�1.02%���ɼ�������������������������С��������ˣ����ϵ缫�ں�������24 h��ﵽ���ȶ���λ���ȴ�Ǧ�缫��Լ100 mV����˵��Pb��������ͬʱ��Ʋ��в�����MnO2�γ���PbO2-MnO2���ϲ㣬�Ծ��нϺõ��������Ч����

ͼ10 ͬһ�缫��������12 hǰ��ı�����ò
Fig.10 Surface morphologies of same anode before (a) and after (b) galvanostatic polarization for 12 h
3 ����
1) ѭ���������Խ��������Pb/Pb-MnO2���������Ϻõ�����������Կ�����Դ��MnO2�������������ã����϶Ʋ�ϴ����ʵ������Լ������������������������������ԸߵĦ�-PbO2��ͬʱ��MnO2�ļ��벢δ�����ı������绯ѧ��Ӧ��Pb�������̣�ֻ��ʹ��������������������ߣ����������á�
2) ����������������������������нϸߵ��������ԣ��ڵ���ʼ����������λ���Ǧ�缫����Լ300 mV��24 h�����ȶ���λ�Ա�Ǧ������100 mV����Pb-Ag(0.6%����������)�������൱�����ǣ����������ĵ�����ܻ��ܵ�Pb���ױ���״̬�仯��Ӱ�죬PbO2��������ʹ����MnO2���䣬�������Լ���������ʵ��ɴӼ�ǿ����Pb��MnO2�Ľ�������֣���������MnO2�������������γ��ȶ���MnO2�����ϸߵ�PbO2-MnO2���ϲ㣬������һ���������������ȶ�������λ��
REFERENCES
[1] ������. пұ��[M]. ��ɳ: ���ϴ�ѧ������, 2005: 103-118.
[2] PENG Rong-qiu. Zinc metallurgy[M]. Changsha: Central South University Press, 2005: 103-118.
[3] ������. Ǧ��Ǧ�Ͻ�[M]. ��ɳ: ���Ϲ�ҵ��ѧ������, 1996: 329-330.
[4] LI Song-rui. Lead and lead alloys[M]. Changsha: Central South University of Technology Press, 1996: 329-330.
[5] FELDER A, PRENGAMAN R D. Lead alloys for permanent anodes in the nonferrous metals industry[J]. JOM, 2006, 58(10): 28-31.
[6] MOATS M S.Will lead-based anodes ever be replaced in aqueous electrowinning?[J]. JOM, 2008, 60(10): 46-49.
[7] LI W S, CHEN H Y, LONG X M. Oxygen evolution reaction on lead-bismuth alloys in sulfuric acid solution[J]. Journal of Power Sources, 2006, 158: 902�C907.
[8] ��ˮƽ, ������, ������, ������, �� ��, ��ҵ��. п����� Pb-Ag-Ca-Sr ��Ԫ�Ͻ�����������������Ϊ[J]. �й���ɫ����ѧ��, 2008, 18(7): 1342�C1346.
[9] ZHONG Shui-ping, LAI Yan-qing, JIANG Liang-xing, TIAN Zhong-liang, LI Jie, LIU Ye-xiang. Anodization behavior on Pb-Ag-Ca-Sr alloy during zinc electrowinning[J]. The Chinese Journal of Nonferrous Metals, 2008, 18(7): 1342�C1346.
[10] IVANOV I, STEFANOV Y, NONCHEVA Z, PETROVA M. Insoluble anodes used in hydrometallurgy. Part ��. Corrosion resistance of lead and lead alloy anodes[J]. Hydrometallurgy, 2000, 57: 109-124.
[11] IVANOV I, STEFANOV Y, NONCHEVA Z, PETROVA M. Insoluble anodes used in hydrometallurgy. Part II. Anodic behavior of lead and lead-alloy anodes[J]. Hydrometallurgy, 2000, 57: 125-139.
[12] CATTARIN S, MUSIANI M. Electrosynthesis of nanocomposite materials for electrocatalysis[J]. Electrochimica Acta, 2007, 52: 2796-2805.
[13] MARSHALL A, BORRESEN B, GAGENM G, TSYPKIN M, TUNOLD R. Electrochemical characterisation of IrxSn1-xO2 powders as oxygen evolution electrocatalysts[J]. Electrochimica Acta, 2006, 51: 3161-3167.
[14] JIRKOVSKY J, MAKAROVA M, KRTIL P. Particle size dependence of oxygen evolution reaction on nanocrystalline RuO2 and Ru0.8Co0.2O2-x[J]. Electrochemistry Communications, 2006, 8: 1417-1422.
[15] LI Bao-song, LIN An, GAN Fu-xing. Preparation and electrocatalytic properties of Ti/IrO2-Ta2O5 anodes for oxygen evolution[J]. Transactions of Nonferrous Metals Society of China, 2006, 16: 1193-1199.
[16] YE Z G, MENG H M, SUN D B. Electrochemical impedance spectroscopic (EIS) investigation of the oxygen evolution reaction mechanism of Ti/IrO2+MnO2 electrodes in 0.5 M H2SO4 solution[J]. Journal of Electroanalytical Chemistry, 2008, 621: 49-54.
[17] SHRIVASTAVA P, MOATS M S. Ruthenium palladium oxide-coated titanium anodes for low-current-density oxygen evolution[J]. Journal of the Electrochemical Society, 2008, 155(7): E101-E107.
[18] HRUSSANOVA A, MIRKOVA L, DOBREV T. Anodic behaviour of the Pb-Co3O4 composite coating in copper electrowinning[J]. Hydrometallurgy, 2001(60): 199-213.
[19] DOBREV T, VALCHANOVA I, STEFANOV Y, MAGAEVA S. Investigations of new anodic materials for zinc electrowinning[J]. Transactions of the Institute of Metal Finishing, 2009, 87(3): 136-140.
[20] ��־��, ���ҳ�, �˾���, ����. Al/Pb-WC-ZrO2-Ag��Al/Pb-WC-ZrO2-CeO2���ϵ缫���ϵ������о�[J]. ����������ѧѧ��: ������, 2007, 32(3): 13-18.
[21] CHANG Zhi-wen, GUO Zhong-cheng, PAN Jun-yi, XU Rui-dong. Property studies on electrodeposited Al/Pb-WC-ZrO2- Ag and Al/Pb-WC-ZrO2-CeO2 composite electrode materials[J]. Journal of Kunming University of Science and Technology: Science and Technology, 2007, 32(3): 13-18.
[22] SCHMACHTEL S, TOIMINEN M. New oxygen evolution anodes for metal electrowinning: MnO2 composite electrodes[J]. J Appl Electrochem, 2009, 39(10): 1835-1848.
[23] ����ͩ, ����Ԫ. ���ϵ�Ƽ���[M]. ����: ��ѧ��ҵ������, 2007.
[24] GUO He-tong, ZHANG San-yuan. Composite plating technology[M]. Beijing: Chemical Industry Press, 2007.
[25] ZAHAVI J, HAZAN J. Electrodeposited nickel composites containing diamond particles[J]. Plating and Surface Finishing, 1983, 70(2): 57-61.
[26] ������. ұ���豸����������ԭ������������[M]. ��ɳ: ���ϴ�ѧ������, 2006: 6-18.
[27] TANG Mo-tang. Metallurgical equipment basic-transfer principle and material transport[M]. Changsha: Central South University Press, 2006: 6-18.
[28] ����ͩ, ������. �绯ѧ�̳�[M]. ���: ����ѧ������, 2005: 115-148.
[29] GUO He-tong, QIN Qi-xian. Electrochemistry tutorial[M]. Tianjin: Tianjin University Press, 2005: 115-148.
[30] �� ��. �������̼������������ľ���ṹ���Ʊ����ŵ�����(1)[J]. ���, 2004, 34(6): 411-414.
[31] XIA Xi. Crystal structure, preparation and discharge performance for manganese dioxides and related manganese oxides(1)[J]. Battery, 2004, 34(6): 411-414.
[32] FERNANDES J B, DESAI B D, KAMAT V N. Studies on chemically precipitated Mn (��) oxides-��. Correlation among physical, chemical and electrochemical characteristics of manganese dioxides[J]. Electrochemica Acta, 1984, 29(2): 187-931.
[33] CELIS J P, ROOS J R, BUELENS C. A mathematical model for the electrolytic codeposition of particles with a metallic matrix[J]. J Electrochem Soc, 1987, 1346(6): 1402-1408.
[34] �� ��. �������̵���������ѧ��������绯ѧ���Ե����(4)[J].���, 2006, 36(3): 195-198.
[35] XIA Xi. The relation between chemical, physical properties and electrochemical activity for manganese dioxides(4)[J]. Battery, 2006, 36(3): 195-198.
[36] YAMANOTO Y, FUMINO K, UEDA T. A potentiodynamic study of the lead electrode in sulphuric acid solution[J]. Electrochimica Acta, 1992, 37(2): 199-203.
[37] CZEERWINSKI A, ZELAZOWSKA M, GRDEN M. Electrochemical behavior of lead in sulfuric acid solutions[J]. Journal of Power Sources, 2000, 85: 49-55.
(�༭ ����)
������Ŀ������ʡԺʿ����������Ŀ(06FJ4059)
�ո����ڣ�2010-06-18�������ڣ�2010-09-13
ͨ�����ߣ�����������ʦ����ʿ���绰��0731-88830474��E-mail��csulightmetals07@163.com
[1] ������. пұ��[M]. ��ɳ: ���ϴ�ѧ������, 2005: 103-118.
[2] PENG Rong-qiu. Zinc metallurgy[M]. Changsha: Central South University Press, 2005: 103-118.
[3] ������. Ǧ��Ǧ�Ͻ�[M]. ��ɳ: ���Ϲ�ҵ��ѧ������, 1996: 329-330.
[23] ����ͩ, ����Ԫ. ���ϵ�Ƽ���[M]. ����: ��ѧ��ҵ������, 2007.
[26] ������. ұ���豸����������ԭ������������[M]. ��ɳ: ���ϴ�ѧ������, 2006: 6-18.
[28] ����ͩ, ������. �绯ѧ�̳�[M]. ���: ����ѧ������, 2005: 115-148.
[30] �� ��. �������̼������������ľ���ṹ���Ʊ����ŵ�����(1)[J]. ���, 2004, 34(6): 411-414.
[34] �� ��. �������̵���������ѧ��������绯ѧ���Ե����(4)[J].���, 2006, 36(3): 195-198.
