
���±�ţ�1004-0609(2015)12-3319-08
Mg-Al-Y�Ͻ���Al-Y�����仯��������ȶ��ԡ����Ժ�����ѧ���ʵĵ�һԭ������
�º��٣��� ����ëƼ���� ��
(������ҵ��ѧ ���Ͽ�ѧ�빤��ѧԺ������ 110870)
ժ Ҫ��
ͨ�������ܶȷ������۵ĵ�һԭ�����㷽������Mg-Al-Y�Ͻ��е���Ҫǿ���࣬��Al2Y��Al3Y�����ȶ��ԡ����ӽṹ�����������Լ�����ѧ���ʽ��м��㡣�������ȵļ�����������Al2Y��Al3Y�����ȶ����ڣ�Al2Y�Ľṹ�ȶ��Ը�ǿ����ˣ��ںϽ�����̹����У�Al2Y����������Al2Y��Al3Y�ĵ���̬�ܶ�(DOS)�Ͳ�ֵ���ܶȼ���Ľ��������Al2Y��Al3Y��������ȶ����ڵ����ڱ�������Alԭ����Yԭ�ӵļ۵��ӹ������ǿ�ҵ�����ã��γ���spd�ӻ��������ڵ�ԭ�ӳɼ���Ϊ���ۼ������Ӽ��ͽ���������ģ��B������ģ��G������ģ��E�����ɱȦͺ�����������A����ѧ���ʲ����ļ�������������������ΪǿӲ�Ĵ����ಢ��Ϊ����ͬ�ԣ���ˣ��������Ƶ�ǿ��Ч���������۵�ϸ߱�������кܺõ����ȶ��ԣ��ܹ���ߺϽ�ĸ������ܡ�����������̬�ܶȼ����Լ��°��¶ȵļ�������һ����֤��������нṹ�ȶ��Խϸߡ����������ѧ���ʷ���һ������ѧ���ɣ����������ܵļ�����������������ȶ���˳��û�з����仯�������¶ȵ����ߣ�Al2Y�Ľṹ�ȶ�����ǿ��Al3Y�ġ�
�ؼ��ʣ�
Mg-Al-Y�Ͻ�����һԭ�����������ȶ��������ӽṹ������������ѧ������
��ͼ����ţ�TG146.2 �� �� ���ױ�־�룺A
First principles calculation of phase stability, elastic and thermodynamic properties of Al-Y intermetallics in Mg-Al-Y alloy
CHEN Hong-lei, LIN Li, MAO Ping-li, LIU Zheng
(School of Materials Science and Engineering, Shenyang University of Technology, Shenyang 110870, China)
Abstract: Phase stability, electronic structure, elastic and thermodynamic properties of Al2Y and Al3Y phases were investigated by means of first�Cprinciples calculations based on density functional theory (DFT). The results of formation enthalpy show that both two phases can stay stably, and Al2Y is more stable than Al3Y. Therefore, Al2Y has the priority to precipitate in the process of alloy solidification. The calculated density of state (DOS) and the electron density difference indicate that the phase stability of Al2Y and Al3Y is due to the strong interaction between valence electron orbits of Al and Y atoms, forming the spd hybridization. The bonding characteristics of Al2Y and Al3Y phases are all covalent bond, ionic bond and metallic bond. The calculated results of bulk modulus (B), shear modulus (G), elastic modulus (E), Poisson��s ratio (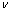 ) and anisotropic coefficient (A) show that Al2Y and Al3Y phases are strong, hard and isotropic. Their similar mechanical properties contribute to their similar performance in Mg-Al-Y alloy. The phonon spectrum, phonon density of states and Debye temperature show their good structural stability as before. High melting points of Al2Y and Al3Y show that their strong thermal stability can improve the mechanical properties of alloy at high temperature. The thermodynamic properties of the two phases conform to the general laws, and the calculated free energy shows that the stability sequence of the two phases has not changed. With the increase of temperature, the structure stability of Al2Y is still better than that of Al3Y.
) and anisotropic coefficient (A) show that Al2Y and Al3Y phases are strong, hard and isotropic. Their similar mechanical properties contribute to their similar performance in Mg-Al-Y alloy. The phonon spectrum, phonon density of states and Debye temperature show their good structural stability as before. High melting points of Al2Y and Al3Y show that their strong thermal stability can improve the mechanical properties of alloy at high temperature. The thermodynamic properties of the two phases conform to the general laws, and the calculated free energy shows that the stability sequence of the two phases has not changed. With the increase of temperature, the structure stability of Al2Y is still better than that of Al3Y.
Key words: Mg-Al-Y alloy; first principles calculation; phase stability; electronic structure; elastic property; thermodynamic property
þ�Ͻ�����ܶ�С����ǿ�ȸߡ��������������ڻ������õ��ص㣬��������þ�Ͻ�չ��ΪѸ�١����ھ������������ṹ���̲�����������ŵ㣬þ�Ͻ��ں��պ��졢���������ӵȹ�ҵ����õ�Խ��Խ�㷺��Ӧ��[1-3]��Mg-Alϵ�Ͻ���Ӧ����Ϊ�㷺��þ�Ͻ�ϵ��֮һ�������������������ܣ�������Ҫ�������-Mg17Al12�����ȶ����ܽϲ��ˣ�������������µ�ʹ�á�ͨ�����ӺϽ�Ԫ�أ��γ����ȶ��Ըߵĵڶ������������ߺϽ�ĸ�����ѧ���ܡ�
�����о�������ϡ��Ԫ�ؿ�ͨ������ǿ��������þ�Ͻ���ۺ���ѧ����[4-6]�����У�Y�dz��õ�ϡ��Ԫ��֮һ������Mg-Al�Ͻ���������Al�����γɶ���Al-Y�����仯���һЩ�о�����Al2Y����Ϊ��Ҫǿ���࣬�ɲ���������ǿ������[7-8]�����⣬�ںϽ�Ŀ������̹����У�Al������Y�����γ�����������Al3Y�����е��ܶȡ��߿������ԡ����۵��Լ����õĸ���ǿ�ȵ��ص㣬������ܹ�ע[9]��
Ŀǰ�������⾡���Ѿ���Mg-Al-Yϵ�Ͻ�����㹻�����ӣ�����չ�˺ܶ�г�Ч���о�������Щ�о���Ҫ�����ںϽ������֯����ѧ���ܷ��棬ֻ�����������ӵ��Ӳ�ζ�Al2Y��Al3Y������˽�Ϊ������о������磬DUAN��[8]Ӧ�õ�һԭ������ķ�����Al3Y������˵������ʵ��о����о���ѹ���Ե������ʵ�Ӱ�죻TAO��[10]������Al2RE������������ж���ṹ�ȶ��ԣ����Ե��Գ��������˼��㡣������Щ�о���Ȼ���뵽�˵��ӽṹ���۲�Σ�����û����Mg-Al-Yϵ�Ͻ���ϵ��ϵ������ֻ�ǹ�������Al2Y��Al3Y����Ϊ�о�Ŀ�꣬���������һЩ������������ʣ��Լ���������������Ⱥͽṹ�ȶ��ԡ�Ϊ�ˣ��������߽�ͨ����һԭ���ķ�������Al2Y��Al3Y�Ľṹ�����ʽ���ȫ�桢ϵͳ������ط�����ͨ������Al2Y��Al3Y�����ȶ��ԡ����ӽṹ�����������Լ�����ѧ���ʣ�����Al2Y��Al3Y���������ʽ��бȽϣ��������϶�������ǿ����Ľṹ�ȶ��ԡ��ɼ����ԣ��Լ���Mg-Al-Yϵ�Ͻ��п��ܾ��е�ǿ�����Ƶȸ����ֵķ��������ۣ�������Mg-Al-Yϵ�Ͻ����ƿ����ṩһ��������ָ����
1 ���㷽��
���û����ܶȷ������۵ĵ�һ��ԭ��ƽ�沨ٞ�Ʒ���[11-12]��CASTEP��������м��㡣���������������ù����ݶȽ���(GGA)[13]�е�PBE(Perdew-Burke- Ernzerhof)[14]������Ǣ����(SCF)�������м��㣬���BFGS�����ݶȷ���������ʵ��۵���֮�������ò��ó���ٞ��[15]��Al��Y�����������Ų��ֱ�Ϊ3s2��4d15s2���ض�������Ϊ340 eV������ģ�͵ļ�Լ����Ԩ����K���������Monkhorst-Pack[16]������K����������Ϊ5��5��5��ÿ��ԭ���ϵ�������0.01 eV/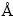 ������ƫ��С��5.0��10-4 ��Ӧ��ƫ��С��0.2 GPa�������Ż��ڵ��ӳ�ԥ�½��У�ֱ��������ֵ�ﵽ1.0��10-8 eV/atom��
������ƫ��С��5.0��10-4 ��Ӧ��ƫ��С��0.2 GPa�������Ż��ڵ��ӳ�ԥ�½��У�ֱ��������ֵ�ﵽ1.0��10-8 eV/atom��
2 ���������
2.1 ����ṹ��������
Al2Y��Al3Y�ľ���ṹ��ͼ1��ʾ������ṹ�����;��������ڱ�1�ͱ�2���������õľ�������ʵ��ֵ�Ƚ��Ǻϣ��������㷽���ǿɿ��ġ�
��������ȼ������ʽ(1)��ʾ��
 (1)
(1)
ʽ�У���HΪ��������ȣ�EtotΪAB�����仯�������������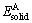 ��
��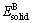 �ֱ�ΪA��Bƽ��ÿ��ԭ�ӵ�������NA��NBΪ������A��Bԭ�ӵĸ���������������Ƚ�����ڱ�3���ӱ�3�п��Կ�����Al2Y��Al3Y����������ֵΪ��ֵ����������������γɹ��̷����ҿ����ȶ����ڡ����һ����ĵ�������Խ�ͣ���������Ԫ�ؼ�ĺϽ�����Խǿ�������γɵ���Խ�ȶ���Al2Y�������ȵ���Al3Y�ģ���˽ṹ�����ȶ����ںϽ�����̹����л��������������о��ļ���ֵ��ʵ��ֵ���ϽϺã������˼��㷽��ȷ�ɿ���
�ֱ�ΪA��Bƽ��ÿ��ԭ�ӵ�������NA��NBΪ������A��Bԭ�ӵĸ���������������Ƚ�����ڱ�3���ӱ�3�п��Կ�����Al2Y��Al3Y����������ֵΪ��ֵ����������������γɹ��̷����ҿ����ȶ����ڡ����һ����ĵ�������Խ�ͣ���������Ԫ�ؼ�ĺϽ�����Խǿ�������γɵ���Խ�ȶ���Al2Y�������ȵ���Al3Y�ģ���˽ṹ�����ȶ����ںϽ�����̹����л��������������о��ļ���ֵ��ʵ��ֵ���ϽϺã������˼��㷽��ȷ�ɿ���
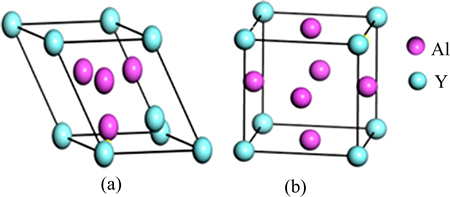
ͼ1 Al2Y��Al3Y�ľ���ṹģ��
Fig. 1 Crystal structure models of Al2Y(a) and Al3Y(b)
��1 Al2Y��Al3Y�ľ���ṹ����
Table 1 Crystal structure parameters of Al2Y and Al3Y
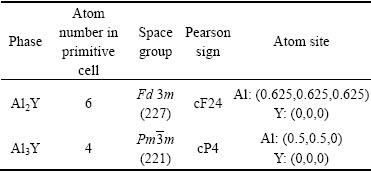
��2 Al2Y��Al3Y��ƽ�⾧����(a)��������ʼ���(V0)���ܶ�(��)
Table 2 Equilibrium crystal parameters(a), unit cell volume(V0) and density(��) of Al2Y and Al3Y
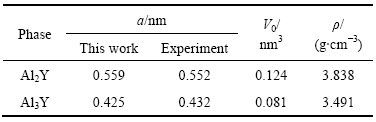
��3 Al2Y��Al3Y��������(��H)
Table 3 Formation enthalpy (��H) of Al2Y and Al3Y
2.2 ���ӽṹ
������Al2Y��Al3Y�ĵ��ӽṹ�Խ�һ���˽���ɼ����ԣ����ҳ����ܹ��ȶ����ڵı���ԭ��Al2Y��Al3Y����̬����̬����̬�ܶ�ͼ��ͼ2��ʾ������Al2Y��Al��s��p�����Լ�Y��s��p��d���Ӿ��Է����ܼ������ijɼ����й��ף���-10~0 eV�ĵ�����Χ�ڣ��ɼ�����Ҫ������Al��s��p���Ӻ�Y��p���ӵĹ���ӻ����ã���0~10 eV�ļ۴���Χ�ڣ��ɼ�����Ҫ������Al��s���Ӻ�Y��p��d���ӵĹ���ӻ����á�����Al3Y����-10~10 eV�ķ����ܼ��������ɼ����ΪAl��p���Ӻ�Y��d���������ף��������ܼ��ĵ��ӹ�������Y�����d���ӽṹ����ijɼ�Ӱ��ܴ�Al2Y��Al3Y�ĵ�����̬�ܶȽ��жԱȿ��Կ������ڷ����ܼ�������Al3Y������ռ�ݵ��ܼ���ΧҪ����Al2Y�ġ���ˣ��ӵ��Ӳ����ϣ�Al2Y�Ľṹ�ȶ���Ҫ����Al3Y�ģ���֮ǰ�����ȵļ�����һ�¡�
Ϊ��һ��ֱ�۵Ľ�ʾ��ijɼ�������������Al2Y��Al3Y����������(111)�IJ�ֵ���ܶ�ͼ����ͼ3��ʾ��Al2YΪAB2�͵�Laves�࣬һ����ԣ�Aԭ��֮����Ҫ�ǽ�������Bԭ��֮����Ҫ�ǹ��ۼ���A��Bԭ��֮����Ҫ�����Ӽ�����ͼ3(a)���Կ��������Ӵ�Alԭ����Yԭ��ת�ƣ�Alԭ��ʧ���ӣ�Yԭ�ӵõ��ӡ�Yԭ��֮��Ϊ��������ϣ�Yԭ�Ӻ����ٽ���Alԭ��֮��Ϊ���Ӽ���ϡ���ͼ3(b)���Կ�����Yԭ�Ӻ����ٽ���Alԭ��֮��Ϊ���ۼ�����Ҿ��н�ǿ�ļ��ԡ�Alԭ��֮��Ϊ��������ϡ�Al2Y��������ԭ��֮��ļ��������Ӽ�Ϊ����Al3Y��������ԭ��֮��ļ����Թ��ۼ�Ϊ���������������ۣ�Al2Y��Al3Y��ԭ�Ӽ�ɼ�������Ҫ�ǹ��ۼ������Ӽ��ͽ���������ˣ�Al2Y��Al 3Y���кܺõĽṹ�ȶ��ԣ��ܹ��ȶ����ڡ�
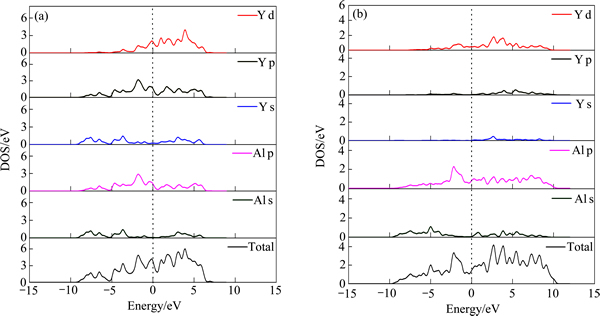
ͼ2 Al2Y��Al3Y����̬����̬����̬�ܶ�
Fig. 2 Total and partial electronic density of state (DOS) of Al2Y (a) and Al3Y (b)
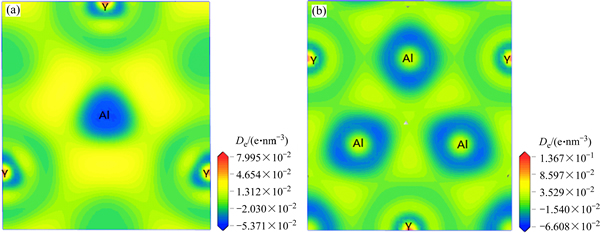
ͼ3 Al2Y��Al3Y�IJ�ֵ���ܶ�(De)
Fig. 3 Electronic density difference (De) of Al2Y (a) and Al3Y (b)
2.3 ��������
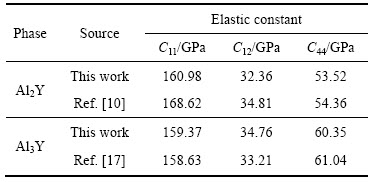
���Գ��������������ϵĵ��ԣ��Dz������������������֮һ���������ṹ���壬����3�������ĵ��Գ���C11��C12��C44���ȶ���������ϵ���Գ�����������������[19]��C11-C12��0��C11��0��C44��0��C11+2C12��0���������õĵ��Գ���ֵ���4���С����μ�����ʵ��ֵ�ǺϽϺã������������Ͳ��������ǿɿ��ġ�Al2Y��Al3Y�ĵ��Գ��������ȶ�����������˿����ȶ����ڡ�
ͨ�����Գ�������ֵ���Լ���������ģ��B������ģ��G������ģ��E�����ɱ�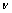 ������������A�����㷽����ʽ(2)~(6)[20]��ʾ��
������������A�����㷽����ʽ(2)~(6)[20]��ʾ��
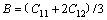 (2)
(2)
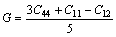 (3)
(3)
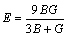 (4)
(4)
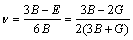 (5)
(5)
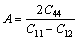 (6)
(6)
�����������ڱ�5�����У���ģ��B�������������Ӧ�������µֿ����ε�����������ֵԽ�ֿ����ε�����Խǿ������ģ��G���������ڼ���Ӧ�������µֿ���Ӧ�������������ֵԽ�����е�����Խǿ[21]������ģ��E�Ͳ��ɱ��ֱ��ʾ���ϵ�Ӧ����Ӧ��ȼ����ϵĿ���������������ģ��Խ������ϵ�����Խ�ã������ɱ�Խ������ϵ�����Խ��[19]����ֵһ��Ϊ-1~0.5��
PUSH[21]��Ϊ������ģ�������ģ��֮��(G/B)�ܹ���ӳ�����ϱ��εĴ����������ص㣬���еļ���ģ�������ģ���ֱ����ֲ��Ͽ����Ա��μ������Զ��ѵ����������ٽ�ֵһ��ָ����0.5����G/B��0.5ʱ�����ϳʴ��Զ��ѣ���G/B��0.5ʱ�����ϳ����Զ��ѡ�(C11-C12)ֵԽС����������Խ�ã�(C12-C44)�������ϵ���չ��(����)����ֵΪ������������Ϊ�����ࣻ��ֵΪ����������Ϊ������[22-23]��
��4 Al2Y��Al3Y�ĵ��Գ���
Table 4 Elastic constants of Al2Y and Al3Y
��5 Al2Y��Al3Y������ѧ����
Table 5 Thermodynamic properties of Al2Y and Al3Y
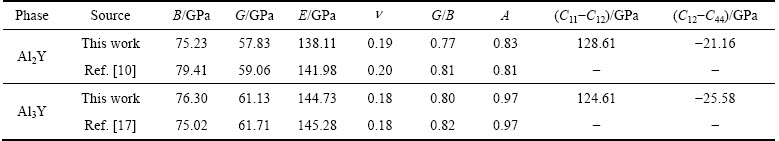
�ӱ�5�п��Կ�����Al2Y��Al3Y��B��G��E��ֵ����������������ܲ������Ƶ�ǿ��Ч�����ϴ��ģ��ֵ������������ΪǿӲ�Ĵ����ࡣ��G/Bֵ������0.5���������߾�Ϊ�����࣬�ӵ��Գ����IJ�ֵ(C11-C12)��(C12-C44)Ҳ���Ա�����һ���ʡ�(C11-C12)��Ϊ�ܴ������������������Խϴ�(C12-C44)Ϊ��ֵ��Ҳ���������Ϊ�����ࡣ���ߵĸ�����������Aֵ��1�ӽ�����ˣ����������ڸ���ͬ�ԡ���Al2Y��Al3Y�ܹ�ϸС��ɢ�طֲ���Mg-Al-Yϵ�Ͻ�����У����ԺϽ���������ǿ�����ã�����˺Ͻ����ѧ���ܡ�
2.4 ����ѧ����
�þ���ṹ�ĵ��Գ��������ģ�����Խ��ƹ��ƶ�Ӧ�����仯������۵�(Tm)������㹫ʽ[24]���ηֱ���ʽ(7)��(8)��ʾ��
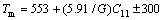 K (7)
K (7)
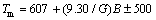 K (8)
K (8)
��Ӧ���۵��������ͼ4��ʾ������ֵ����������ǿ�����Ϊ���۵��࣬���������ǿ�����ڸ������ܹ��ȶ����ڣ��Ӷ���������˺Ͻ�ĸ������ܡ�
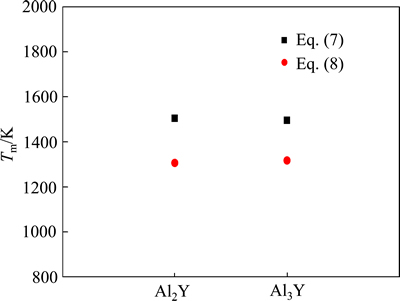
ͼ4 Al2Y��Al3Y���۵����ֵ
Fig. 4 Theoretically calculated Tm of Al2Y and Al3Y through Eq.(7) and Eq.(8)
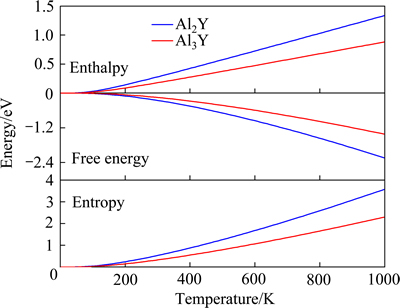
ͼ5 Al2Y��Al3Y���ʡ��ء����������¶�(0~1000 K)�ı仯
Fig. 5 Enthalpy, free energy and entropy of Al2Y and Al3Y as a function of various temperatures (0-1000 K)
�������Ӽ�������������۽���г��������ʡ��ء������ܡ��������¶ȵ�������ϵ�Լ��°��¶ȡ�ͼ5��ʾΪ����õ���Al2Y��Al3Y���ʡ��ء����������¶ȵı仯���ߡ���0~1000 K���¶������ڣ����������û�н��㣬����Al2Y�����ȶ������Ǹ���Al3Y�ģ���֮ǰ�Ľ�����һ�£�ʹ��0 K�µļ����������ƴ������µļ������ǿ��еġ�
����BORN��[25]�ĵ����ȶ��оݣ��ȶ����������Ƶ��Ϊʵ��������Ƶ��Ϊ��������������ζ�Ŷ�Ӧ��ģ�Ļָ��������ڻ�̫С������������ڸ���ģʽ��ʧ�ȡ�Al2Y��Al3Y������ɫɢͼ������̬�ܶ���ͼ6��ʾ����ͼ6���Կ�����Al2Y��Al3Y������Ƶ��Ϊʵ�����������߾�Ϊ�ȶ����壬����ѧ�ȶ��ṹ��
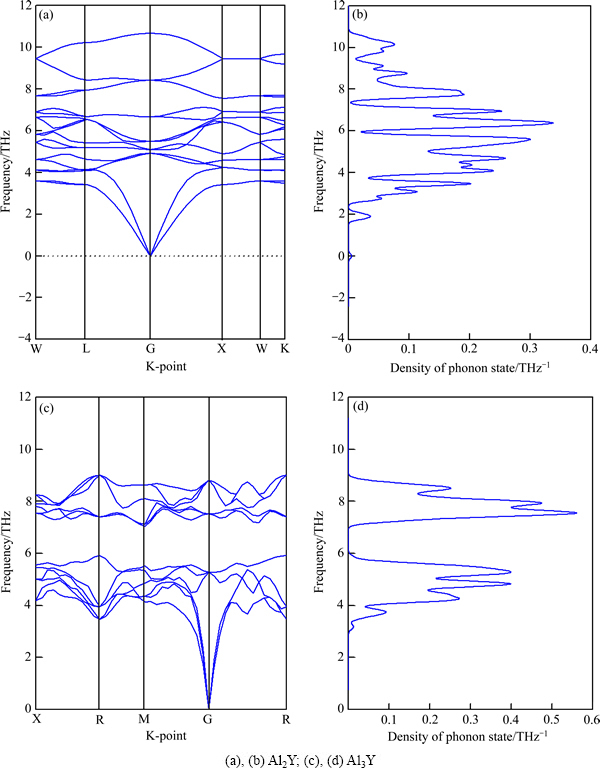
ͼ6 Al2Y��Al3Y������ɫɢͼ������̬�ܶ�
Fig. 6 Phonon spectra((a), (c)) and phonon density of states((b), (d)) of Al2Y and Al3Y
�ڼ����˾���ĵ��Գ����Լ���ѧ����֮�������ۼ���������µĵ°��¶ȡ��°��¶�Խ�ߣ�����Ĺ��ۼ�ǿ��Խǿ[26]��������ṹ�������Ե��ȶ��ԡ�Al2Y��Al3Y�ĵ°��¶�(TD)��ͨ���ྦྷ���ƽ�����Ӳ���(vm)��ʽ(9)����[27-28]��
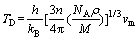 (9)
(9)
ʽ�У�hΪ���ʿ˳�����kBΪ��������������NAΪ����٤��������nΪ�����е���ԭ��������Ϊ�ܶȣ�MΪĦ���������ྦྷ���ƽ�����Ӳ���(vm)������ʽ(10)���㣺
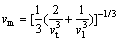 (10)
(10)
ʽ�У�vl��vt�ֱ����ݲ��ͺᲨ���٣�����ͨ��Navier��s ��������ģ��B�ͼ���ģ��G����õ�[29]��
 (11)
(11)
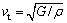 (12)
(12)
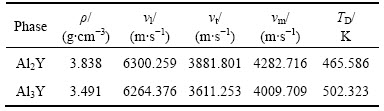
Al2Y��Al3Y���ݲ��ͺᲨ����vl��vt��ƽ������vm�͵°��¶�TD�����۽�����ڱ�6��������ĵ°��¶���ֵ��ʵ��ֵ����������ֵ473 K[30]��503 K[17]���ϽϺá�����ĵ°��¶Ⱦ��ϸߣ���������������ԭ��֮��Ĺ����Խ�ǿ����������ṹ�����ȶ�����֮ǰ�����������ݺ͵���̬�ܶ�(DOS)�Ľ���һ�¡�Al3Y�ĵ°��¶ȸ���һЩ��˵��Al3Y����ԭ�ӹ����Ը���Al2Y�ģ������ֵ����ܶȵĽ��������
��6 Al2Y��Al3Y��ѧ���ݵ����ۼ���ֵ
Table 6 Theoretically calculated thermal properties of Al2Y and Al3Y phases
3 ����
1) ��Mg-Al-Y�Ͻ��е�Al2Y��Al3Y����е�һԭ���ļ���ģ�⣬���õ�ƽ�⾧������Լ������ȵ���ֵ��ʵ��ֵ����������ֵ��һ�£�Al2Y�Ľṹ�ȶ��Ը���Al3Y�ģ��Ͻ�������ǿ��
2) ���ӽṹ�ļ��������Al2Y��Al3Y�ijɼ�����Ϊ���ۼ������Ӽ��ͽ�������Al2Y����ԭ��֮��Ϊ���Ӽ���Al3Y����ԭ��֮��Ϊ���ۼ���
3) Al2Y��Al3Y����ѧ���ʺ����ƣ���ΪǿӲ�Ĵ����࣬�������Ƶĵڶ���ǿ��Ч����
4) Al2Y��Al3Y���۵���ϸߣ��ڸ����¿����ȶ����ڣ�������ߺϽ�ĸ������ܡ����Ӽ���Ҳ��������������ȶ����ڡ�����ĵ°��¶�Ҳ�ϸߣ���ԭ�Ӽ�ļ��Ͻ�ǿ�����ȶ��Ժá�
REFERENCES
[1] �� ��, �� ��, ��־÷, ����ʯ, ������. Mg-Gd��Ԫ�Ͻ��Ц¡�-Mg7Gd������ĵ�һ��ԭ�������о�[J]. ��ѧͨ��, 2010, 30: 2968-2973.
GAO Lei, ZHOU Jian, SUN Zhi-mei, CHEN Rong-shi, HAN En-hou. First-principles calculations of the �¡�-Mg7Gd precipitate in Mg-Gd binary alloys[J]. Chin Sci Bull, 2010, 30: 2968-2973.
[2] ֣ΰ��, ��˫��, �� ��, ����. ���ϡ����AZ91Dþ�Ͻ���֯����ѧ���ܵ�Ӱ��[J]. ����ѧ��, 2006, 42(8): 835-842.
ZHENG Wei-chao, LI Suang-shou, TANG Bin, ZENG Da-ben. Effects of mischmetal on microstructure and mechanical properties of AZ91D magnesium alloy[J]. Acta Metal Sinica, 2006, 42(8): 835-842.
[3] �� ��, ���ζ�, �ź��, �� ��, �γ���. ϡ����þ�Ͻ������õ��о���״[J]. ���ϵ���, 2011, 25(17): 487-491.
WANG Yong, LIAO Zhi-dong, ZHANG Heng-fei, ZHOU Hong, SONG Chang-jiang. Development on effects of rare earth element in magnesium alloys[J]. Mater Rev, 2011, 25(17): 487-491.
[4] JUNG I, SANJARI M, KIM J, YUE S. Role of RE in the deformation and recrystallization of Mg alloy and a new alloy design concept for Mg�CRE alloys[J]. Scripta Mater, 2015, 102: 1-6.
[5] LUKYANOVA E A, ROKHLIN L L, TABACHKOVA Y N, DOBATKINA T V, NIKITINA N I. Reversion after ageing in an Mg-Y-Gd-Zr alloy[J]. J Alloys Compd, 2015, 635: 173-179.
[6] ������, ������. ϡ��Ԫ��������þ�Ͻ���Ӧ�õ��о���״���䷢չ����[J]. �й�ϡ��ѧ��, 2010, 28(6): 643-653.
XIN Ming-de, JI Ze-sheng. Research situation and application prospects of rare earth in foundry magnesium alloy[J]. J Mater Sci Technol, 2010, 28(6): 643-653.
[7] LUO A, PEKGULERYUZ M O. Cast magnesium alloys for elevated temperature applications[J]. J Mater Sci, 1994, 29: 5259-5271.
[8] DUAN Y H, SUN Y, PENG M J, ZHOU S G. Ab-initio investigations on elastic properties in L12 structure Al3Sc and Al3Y under high pressure[J]. J Alloys Compd, 2014, 585: 587-593.
[9] HARADA Y, DUNAND D C. Creep properties of Al3Sc and Al3(Sc, X) intermetallics[J]. Acta Mater, 2000, 48: 3477-3487.
[10] TAO X M, OUYANG Y, LIU H, FENG Y P, DU Y, JIN Z P. Ab initio calculation of the total energy and elastic Properties of Laves Phase C15 A12RE (RE=Sc,Y,La,Ce-Lu)[J]. Comp Mater Sci, 2008, 44: 392-399.
[11] �ջԽ�, �� ��, ��־��, �Ŵ���, �� ��, �Ʋ���. ����Mg�����ȶ��Եĵ�һԭ�����ӽṹ����[J]. �й���ɫ����ѧ��, 2008, 18(12): 2224-2232.
TAO Hui-jin, YIN Jian, YIN Zhi-min, ZHANG Chuan-fu, LI Jie, HUANG Bai-yun. Calculations of lattice stabilities of elemental Mg from electronic structures in first principles[J]. The Chinese Journal of Nonferrous Metals, 2008, 18(12): 2224-2232.
[12] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review, 1990, 41: 7892-7895.
[13] WHITE J A, BIRD D M. Implementation of gradient-corrected exchange-correlation potentials in Car-Parrinello total-energy calculations[J]. Physical Review, 1994, 50: 4954-4957.
[14] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation exchange-correlation energy[J]. Phys Rev Lett, 1996, 77: 3865-3868.
[15] FRANCIS G P, PAYNE M C J. Finite basis set corrections to total energy pseudopotential calculations[J]. J Phys Condens Mat, 1990, 2: 4395-4404.
[16] ��˳ƽ, ��Сƽ, �� �S, ¬����, � ��, ����, �� ��. L12-Al3Li�����仯�����ȱ��Ũ�ȵĵ�һԭ������[J]. �й���ɫ����ѧ��, 2013, 23(2): 370-378.
SUN Shun-ping, LI Xiao-ping, YU Yun, LU Ya-lin, ZANG Bing, YI Dan-qing, JIANG Yong. First-principle calculation of point defects concentration in L12-Al3Li intermetallic[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(2): 370-378.
[17] ��С��. ϡ������þ�Ͻ�����ѧ���ʵĵ�һԭ������[D]. ��ɳ: ���ϴ�ѧ, 2008.
TAO Xiao-ma. First-Principles calculations of the thermodynamic properties of rare earth-aluminum and rare earths-magnesium alloys[D]. Changsha: Central South University, 2008.
[18] LIU S, DU Y, XU H, HE C, SEHUSTER J C. Experimental investigation of the Al-Y phase diagram[J]. J Alloys Compd, 2006, 414: 60-65.
[19] LIU Q, ZHANG R. Effect of 6.25 at% Al addition on structural stability of magnesium under high pressure: A first-principles study[J]. J Alloys Compd, 2010, 508(2): 616-619.
[20] HILL R. The elastic behaviour of a crystalline aggregate[J]. Phys Soc Sect A, 1952, 65: 349-354.
[21] PUSH S F. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals[J]. Philos Mag, 1954, 45: 823-843.
[22] FU C L, WANG X D, YE Y Y. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallics, 1999, 7: 179-184.
[23] BRUTTI S, NGUYEN-MANH D, PETTIFOR D G, MANFRINETTI P, NAPOLETANO M, CANEPA F. Electronic, electrical and thermodynamic properties of Ca5Si3 by first principles calculations and low temperature experimental techniques[J]. Calphad, 2009, 33: 260-264.
[24] MAO P L, YU B, LIU Z, WANG F, JU Y. Mechanical properties and electronic structures of MgCu2, Mg2Ca and MgZn2 Laves phases by first principles calculations[J]. Transactions of Nonferrous Metals Society of China, 2014, 24(9): 2920-2929.
[25] BORN M, HUANG K. Dynamical theory of crystal lattices[M]. Oxford: Clarendon, 1954.
[26] MORUZZI V L, JANAK J F, SCHWARZ K. Calculated thermal properties of metal[J]. Phys Rev B, 1988, 37: 790-799.
[27] ANDERSON O L. A simplified method for calculating the Debye temperature from elastic constants[J]. J Phys Chem Solids, 1963, 24: 909-917.
[28] GRIMVALL G. Thermo physical properties of materials[M]. North-Holland: Amsterdam, 1986.
[29] WACHTER P, FILZMOSER M, REBIZANT J. Electronic and elastic properties of the light actinide tellurides[J]. Physica B, 2001, 293: 199-223.
[30] HUNGSBERG R E, jr GSCHNEIDNER K A. Low temperature heat capacity of some rare earth aluminum laves phase compounds:YA12, LaA12 and LuA12[J]. J Phys Chem Solids, 1972, 33: 401-407.
(�༭ �� ��)
������Ŀ��2013����ʡ�Ƽ����������Ŀ(2013201018)
�ո����ڣ�2015-04-19�������ڣ�2015-09-21
ͨ�����ߣ��� ���������ڣ���ʿ���绰��13609824337��E-mail: linli-1969@126.com
ժ Ҫ��ͨ�������ܶȷ������۵ĵ�һԭ�����㷽������Mg-Al-Y�Ͻ��е���Ҫǿ���࣬��Al2Y��Al3Y�����ȶ��ԡ����ӽṹ�����������Լ�����ѧ���ʽ��м��㡣�������ȵļ�����������Al2Y��Al3Y�����ȶ����ڣ�Al2Y�Ľṹ�ȶ��Ը�ǿ����ˣ��ںϽ�����̹����У�Al2Y����������Al2Y��Al3Y�ĵ���̬�ܶ�(DOS)�Ͳ�ֵ���ܶȼ���Ľ��������Al2Y��Al3Y��������ȶ����ڵ����ڱ�������Alԭ����Yԭ�ӵļ۵��ӹ������ǿ�ҵ�����ã��γ���spd�ӻ��������ڵ�ԭ�ӳɼ���Ϊ���ۼ������Ӽ��ͽ���������ģ��B������ģ��G������ģ��E�����ɱȦͺ�����������A����ѧ���ʲ����ļ�������������������ΪǿӲ�Ĵ����ಢ��Ϊ����ͬ�ԣ���ˣ��������Ƶ�ǿ��Ч���������۵�ϸ߱�������кܺõ����ȶ��ԣ��ܹ���ߺϽ�ĸ������ܡ�����������̬�ܶȼ����Լ��°��¶ȵļ�������һ����֤��������нṹ�ȶ��Խϸߡ����������ѧ���ʷ���һ������ѧ���ɣ����������ܵļ�����������������ȶ���˳��û�з����仯�������¶ȵ����ߣ�Al2Y�Ľṹ�ȶ�����ǿ��Al3Y�ġ�
[2] ֣ΰ��, ��˫��, �� ��, ����. ���ϡ����AZ91Dþ�Ͻ���֯����ѧ���ܵ�Ӱ��[J]. ����ѧ��, 2006, 42(8): 835-842.
[3] �� ��, ���ζ�, �ź��, �� ��, �γ���. ϡ����þ�Ͻ������õ��о���״[J]. ���ϵ���, 2011, 25(17): 487-491.
[6] ������, ������. ϡ��Ԫ��������þ�Ͻ���Ӧ�õ��о���״���䷢չ����[J]. �й�ϡ��ѧ��, 2010, 28(6): 643-653.
[17] ��С��. ϡ������þ�Ͻ�����ѧ���ʵĵ�һԭ������[D]. ��ɳ: ���ϴ�ѧ, 2008.
[25] BORN M, HUANG K. Dynamical theory of crystal lattices[M]. Oxford: Clarendon, 1954.
[28] GRIMVALL G. Thermo physical properties of materials[M]. North-Holland: Amsterdam, 1986.
