
DOI��10.19476/j.ysxb.1004.0609.2018.08.01
Al-Ti-B�Ͻ���AlB2��TiB2��TiAl3�ĵ�һ��ԭ���о�
��Ԫ��1, 2, 3���ۺ��1��Ф����1, 2, 3������κ1��������1
(1. ���ϴ�ѧ ��Ͻ��о�Ժ, ��ɳ 410083��
2. ���ϴ�ѧ �����ܸ�����������ص�ʵ����, ��ɳ 410083��
3. ���ϴ�ѧ ���繤��ѧԺ, ��ɳ 410083)
ժ Ҫ��
���û����ܶȷ������۵ĵ�һ��ԭ���������о�0 K������Al-Ti-B��ϵ��AlB2��TiB2��TiAl3��ƽ�⾧�������γ��ȡ�����ܼ��������ԣ��������г�°�ģ�Ͷ�3�������ڸ��������µ�����ѧ�������ʽ����˷�����������������AlB2��TiB2��TiAl3���γ��������ܾ�Ϊ��ֵ������3��������нṹ�ȶ��ԣ����ȶ����ɴ�С����ΪTiB2��TiAl3��AlB2��3���������������������ɴ�С����ΪTiAl3��AlB2��TiB2��TiAl3��AlB2��ʾ���Ӽ����ԣ���TiB2���������Թ��ۼ�Ϊ����3����������ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3����������TiB2��AlB2��TiAl3������ѧ���ʿ������ڷ������η��Ʊ�Al-Ti-BʱAl���������֮�䷴Ӧ��������Լ��м�Ͻ����յ�������ɡ�
�ؼ��ʣ�
Al-Ti-B�Ͻ�����һ��ԭ������г�°�ģ�����ṹ����������ѧ������
���±�ţ�1004-0609(2018)-08-1491-08���� ��ͼ����ţ�TG146.2���� ���ױ�־�룺A
��ҵ��Al-Ti-B�ij����Ʊ������ǽ��ߴ������ۻ���ͨ�����벻ͬ�����Ļ�Ϸ���(K2TiF6��KBF4)���������в����·�Ӧһ��ʱ��������Ӷ��õ���ͬTi/B������Al-Ti-B�м�Ͻ�[1-2]��Al-Ti-B�еĵڶ�������(��TiB2��TiAl3��(Al,Ti)B2��)�����������������κ˶��ﵽϸ��������Ч�������磬Al-5Ti-1B������Al������ͷų��ĸ��۵�TiB2���ӿ�����Ϊ��(Al)�����κ˵ĺ��ģ����۵��TiAl3�����������ܱ�Ѹ���ܽⲢ�ͷų�TiԪ�أ�Ϊ��(Al)���������κ˴����˱�������[3-4]����KORI��[5]�о�����Al-1Ti-3B��AlB2�����ϸߣ�����Al-Si�Ͻ�ľ���ϸ��������Al-Ti-B�м�Ͻ����ϸ��Ч�ʸߡ��ɱ��������ŵ������������Ȼ����ʵ��Ӧ�ù������Դ������ڲ��ڶ������Ӿۼ��������ߴ���������ȱ��[6-7]����������Al-Ti-B�ڸ��������������е�Ӧ�á���ˣ������˽���Щ�����κ����ӵĽṹ�����ʾ��Ե���Ϊ��Ҫ��
��ĿǰΪֹ�������о���[8-10]��ͼ�����Ʊ������з������������еķ�Ӧ���Ƽ�������Ը���Al-Ti-B���Ʊ����գ��Ӷ���һ������Al-Ti-B��ϸ��DZ�ܡ�Ȼ����K2TiF6��KBF4��Al�ķ�Ӧ����TiB2��TiAl3���м���Ĺ����ܻ�ѧ��Ӧ���ơ�Ԫ����ɢ���ڶ�����Ӱ�죬���Ծ�ȷ���������з��η�Ӧ�������������߲��û����ܶȷ�������(Density functional theory��DFT)[11-12]�ĵ�һ��ԭ���������о�AlB2��TiB2��TiAl3�Ľṹ�ȶ����Լ��������ԣ��������г�°�ģ�Ͷ���3�������ڸ��������µ�����ѧ�������ʽ��з�����������ԭ�ӽǶȽ�ʾ3��������Al-Ti-B��ϵ�µĻ������ʣ�Ϊ��������Ʒ��Al-Ti-B�Ʊ������ṩ����֧�š�
1 ���㷽����ģ��
�ڽṹ�ȶ�������ӽṹ���о����棬�������߲��û����ܶȷ������۵ĵ�һ��ԭ��ƽ�沨���Ʒ��������ڼ��������ʹ�ù����ݶȽ��� (Generalized gradient approximation, GGA)[13]�������������ܣ����������Ʋ�ȡPerdew-Burke-Ernzerhof (PBE)[14]��ʽ���С���Ԫ�ؼ۵��ӵ�ѡȡ������£�B-2s22p1��Al-3s23p1��Ti-3s23p63d24s2��AlB2��TiB2��TiAl3����ģ�ͼ�Լ����Ԩ��K-point��ȡMonkhorst-Pack[15]�������֣������������ֱ�ȡ10��10��8��10��10��8��8��8��8����Ǣѭ�����������������������Ϊ1��10-6 eV/atom������ԭ��֮��������������0.01 eV/nm������ƫ�Ƶ���5.0��10-4 nm��Ӧ��ƫ��Ϊ����0.05 GPa��
������г�°�ģ��[16]��TiB2��AlB2�Լ�TiAl3�ĵ���Ħ�����ݡ���ֵ�Լ���ƽ�������������ϵ�����¶ȵı仯�������о���3������ķ�ƽ��Gibbs����G*(V; P, T)Ϊ������Ϊ
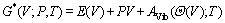 (1)
(1)
ʽ�У�E(V)��ʾ3������ÿ��ԭ������µ���������P��V��T�ֱ�Ϊѹǿ��������¶ȣ���(V)Ϊ�°��¶ȣ�AVibΪ��Helmholtz�����ܡ�
������г���ƺ�ʹ������̬�ܶȵĵ°�ģ�ͣ�AVib������������ʽ��ʾ[17]��
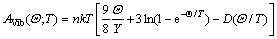 (2)
(2)
ʽ�У�n����ÿ��ԭ��������ԭ����Ŀ��kΪ��������������D(��/T)Ϊ�°ݻ��֡�
����ͬ�Թ���ĵ°��¶Ȧ���ʾ����[16]��
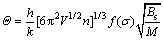 (3)
(3)
ʽ�У�MΪÿ��ԭ���з��ӵ�������Bs�DZ�������ѹ���ʵľ����嵯ģ�������Խ���дΪ
 (4)
(4)
ʽ�У�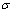 Ϊ���ɱ���(���о���TiB2��AlB2��TiAl3�ֱ�ȡΪ0.117[18]��0.28[19]��0.169[20])����ˣ�ͨ�����������Сֵ��������ƽ�⼪��˹����
Ϊ���ɱ���(���о���TiB2��AlB2��TiAl3�ֱ�ȡΪ0.117[18]��0.28[19]��0.169[20])����ˣ�ͨ�����������Сֵ��������ƽ�⼪��˹����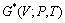 ���Լ�Ϊ����V�ĺ�����
���Լ�Ϊ����V�ĺ�����
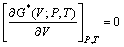 (5)
(5)
ͨ����ⷽ��(5)�����Եõ�״̬����(Equation of state, EOS) V(P, T)��������cV����S�Լ�������ϵ��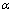 �ֱ���Ա�ʾΪ
�ֱ���Ա�ʾΪ
 (6)
(6)
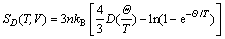 (7)
(7)
 (8)
(8)
ʽ�У�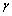 Ϊ���ְ�ɭ(Gruneisen)������
Ϊ���ְ�ɭ(Gruneisen)������
2 ���������
2.1 ����ṹ���ȶ���
TiB2(AlB2)����������ϵ���ռ��ȺΪP6/mmm��ÿ����������1��Ti(Al)ԭ�Ӻ�2��Bԭ�ӣ�Ti(Al)ԭ��ռ���������Ķ��Ǻ͵���λ�ã�����ԭ�Ӵ������ģ�TiAl3�����ķ�����ṹ������I4/mmm�ռ��Ⱥ������������������������Ѷ�ɣ�����2��Tiԭ�Ӻ�6��Alԭ�ӡ�ͨ����ֳ�ԥ�����ṹ�Լ�����ԭ��λ���Դﵽ�������״̬����3������Ľṹ�ȶ��Խ����о�������
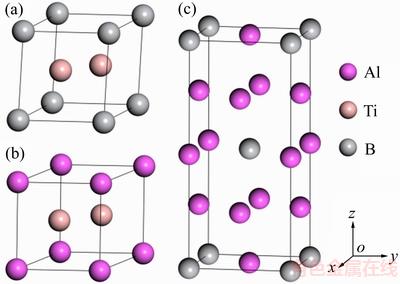
ͼ1 TiB2��AlB2��TiAl3�ľ���ṹģ��
Fig. 1 Structural models for TiB2, AlB2 and TiAl3
�����γ���(Heat of formation, ��H)���Ա��������ɵ���״̬ת��ɾ��廯����״̬�ķ�Ӧ�����������ı仯�������HΪ��ֵʱ����ʾ���������γɹ����з��ȣ���Ӧ���Է����У���֮��Ϊ���ȣ���Ӧ��Ҫ����ṩ�������ܽ��У������ɵĻ����ﲻ�ȶ��������(Cohesive energy, Ecoh)��ָԭ��������̬��ϳ�Ϊ���廯����Ĺ��������ͷŵ����������������������γ�֮���ڽṹ�ϵ��ȶ��ԣ�����ܵľ���ֵԽ�����γɵľ��廯����Խ�ȶ�[21]�����ڻ�����AxBy�����γ��������ܵļ��㹫ʽ�ֱ�����[22]��
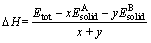 (9)
(9)
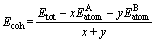 (10)
(10)
ʽ�У�EtotΪ�ṹ�Ż�֮������������� ��
�� �ֱ�Ϊԭ��A��B���ȶ�����״̬ʱ��������
�ֱ�Ϊԭ��A��B���ȶ�����״̬ʱ��������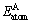 ��
��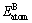 ��ΪA��Bԭ�ӵĹ���̬������x��y�ֱ��ʾ���������ʽ��A��Bԭ�ӵ�ԭ������
��ΪA��Bԭ�ӵĹ���̬������x��y�ֱ��ʾ���������ʽ��A��Bԭ�ӵ�ԭ������
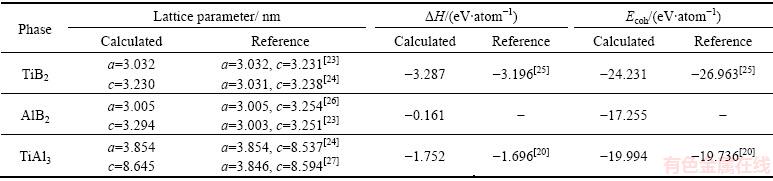
AlB2��TiB2��TiAl3�ľ���ṹ�����Լ��γ��������ܵļ��������1��ʾ��ͨ���ȽϿ��Է��֣����ü�������ʵ�����ֵ����С���������о������õļ���������ú�����
��������ʾ��TiB2��AlB2�Լ�TiAl3���γ��Ⱦ�Ϊ��ֵ������3��������γɹ�����������ṩ�����������Է����У��ӽ���ܵļ�����������3������Ľ���ܾ���ֵ�ɴ�С������ΪTiB2��TiAl3��AlB2��˵��Tiԭ����Bԭ�ӽ��������ǿ�����γɵ�TiB2���۵���ߡ����⣬�Ƚ�����������Է���TiB2�Ľ���ܵ���AlB2��˵����Al-Ti-B�м�Ͻ���TiB2�����ȶ���������[23]���о����������γ��������ܵĽ��������3�ֽ����仯������ȶ����ɴ�С����ΪTiB2��TiAl3��AlB2��
2.2 ���ӽṹ
Ϊ�˽�TiB2��AlB2��TiAl3�ijɼ����ԣ�����һ����ʾ��3������Ľṹ�ȶ��Բ�𣬱��о��зֱ������3���������̬�ܶ�(Total density of state, DOS)�ͷ�̬�ܶ�(Partial density of state, PDOS)�������ͼ2��ʾ��ͼ�����߱�ʾ�����ܼ�(Fermi level)��
��1 ��������TiB2��AlB2�Լ�TiAl3�ľ���ṹ�������γ����������Լ�ʵ��ֵ
Table 1 Calculated and experimental lattice constants, heat of formation and cohesive energy of TiB2, AlB2 and TiAl3
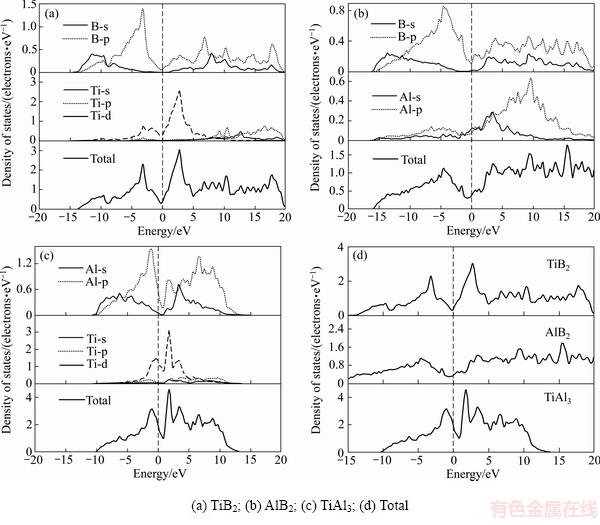
ͼ2 ����ķ�̬�ܶ�����̬�ܶ�
Fig. 2 Partial and total density of states
TiB2��AlB2��TiAl3�ڷ����ܼ�����̬�ܶȾ�����0����ʾ��ͬ�̶ȵĽ������ԡ�ͼ2(a)��ʾTiB2��B 2s��2p��̬�ܶȷ����ص�������sp̬�ӻ���Tiԭ��PDOS��ʾ3d����ڷ����ܼ��������ֳ�������壬����3d����غϵ�s��p����ڷ����ܼ�������̬�ܶȽ�������������Ti 3d��4s��4p���֮����ӻ�����ɵ�[28]��B 2p��Ti 3d���̬�ܶȼ������ڷ����ܼ����Ҷ���ʾ����ǿ���ӻ�����ʹ��TiB2����̬�ܶ��ڷ����ܼ�����������϶����ӳ��TiB2���ڽ�ǿ���ۼ����ԣ�ͼ2(b)��ʾAlB2��B 2s��2p̬�ܶ�ͬ�������ص�������TiB2��ȣ��ص���������Χ��Ϊ�����������ڷ����ܼ�����(0~20 eV)�չ���е�̬�ܶȸ��������AlB2�ķ�����̬���ߡ���һ���棬Alԭ���ڷ����ܼ����´��ڵ�sp̬�ӻ������������ڸ���������ķ�����̬��ǿ����ˣ�Alԭ�ӵļ۵��Ӷ�AlB2�Ĺ��ۼ�����û�й��ס�AlB2����̬�ܶ����ƽ���������ڷ����ܼ�������ƽ̹����϶����ζ�ųɼ�����뷴�����֮������������С�����ۼ��ļ���ǿ�ȱ�TiB2����ͼ2(c)��ʾTiAl3����̬�ܶȼ�����-10~15 eV��������Χ�������ڷ����ܼ�������������϶������TiAl3ͬ������һ���Ĺ��ۼ����ԡ�Ȼ������϶��Χ��խ��˵��TiAl3�Ĺ��ۼ�ǿ�����������������TiAl3��PDOS��֪����̬����Ҫ����Al 3s��3p�Լ�Ti 3d����۵��ӹ���(d�����PDOSΪ�ϴ�ļ�壬�ù��������ԱȽϾ���)�����⣬Al 3p��Ti 3d��PDOS�ڷ����ܼ����������ص�������Al 3s��Ti 3d̬�����ӻ���
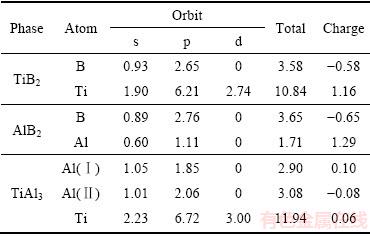
Mulliken��ɲ��ӷ������Զ���������3�������ڲ����ת��������������2��ʾ��
�ӱ�2�п��Կ�����TiB2��Tiԭ��ת�Ƶ�Bԭ�ӵĵ����Ϊ1.16��ת�Ƶĵ����Ҫ����Tiԭ��4s�����AlB2��Alԭ��ת�Ƶ�Bԭ�ӵĵ����Ϊ1.29����TiAl3�г���Al 3s��Ti 3d��������ӻ��������ת��(Tiԭ��ת�Ƶ�Alԭ�ӵĵ����Ϊ0.06)�⣬Al��sp����ӻ�ʹ�õ�λAlԭ��֮�����0.10�ĵ��ת�ơ����Ͻ��������3����������˵��ת�ƣ�ԭ��֮����˹��ۼ�֮�⣬�����������Ӽ����ԣ�����������˳�����ӣ��ɴ�С����ΪTiAl3��AlB2��TiB2��
��2 TiB2��AlB2��TiAl3 ��Mulliken��ɲ���
Table 2 Mulliken electron occupation numbers of TiB2, AlB2 and TiAl3
TiB2��AlB2�Լ�TiAl3�Ľ�����(fm)����ͨ�����¼��㹫ʽ[29]�õ�
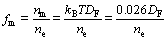 (11)
(11)
ʽ�У�DFΪ�����ܼ���̬�ܶ�ֵ��nm��ne�ֱ�Ϊ�������ȼ�����ӡ��۵��ӵ��ܶȣ���ne���ɾ������(Vcell)���ܼ۵�����(N)����õ���ne=N/Vcell����ز����Լ����������3��ʾ���ɱ�3���Կ�����TiAl3��fm��Խϴ������AlB2����TiB2�Ľ�������С��
��3 ��������ܼ���̬�ܶ�ֵ���ܼ۵���������������Լ������Բ���
Table 3 Density of states at Fermi level (DF), total number of valence electrons (N), cell volume (Vcell) and metallicity parameter (fm) for phases
2.3 ����ѧ����
��������TiB2��AlB2��TiAl3�Ľṹ�ȶ����Լ��������Ծ����ڻ�̬(0 K)�����¼���ó��ġ�Ϊ�˸��ӷ���ʵ��������3������Ļ����������ʣ����߲�����г�°�ģ�ͣ�����GIBBS����[16]��TiB2��AlB2��TiAl3������cV����S�Լ���ƽ�����V��������ϵ�����¶ȵı仯�������о���������Ҫ̽��900~1200 K�¶ȷ�Χ��3��������ȶ���ѧ���ʣ���ͨ����Ҳ��ʵ�ʹ�ҵ�������Ʊ�Al-Ti-B�м�Ͻ���¶ȷ�Χ(700~900��)������õ���ѹ������(300 K)������TiB2��AlB2��TiAl3���嵯ģ��B0�Լ����ѹǿ��һ����B'���4��ʾ���������������о��Լ�ʵ��ֵ���ϽϺá����ڵ�һ��ԭ�����㷽���߹���ԭ�Ӽ������ǿ�ȣ����3����B0�����ۼ���ֵ���Դ���ʵ��ֵ��
��4 TiB2��AlB2��TiAl3���嵯ģ��B0�Լ����ѹǿ��һ����B'
Table 4 Bulk modulus B0 and first derivatives B' of TiB2, AlB2 and TiAl3
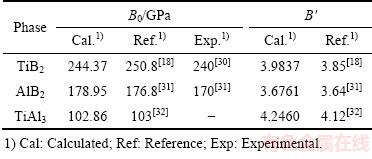
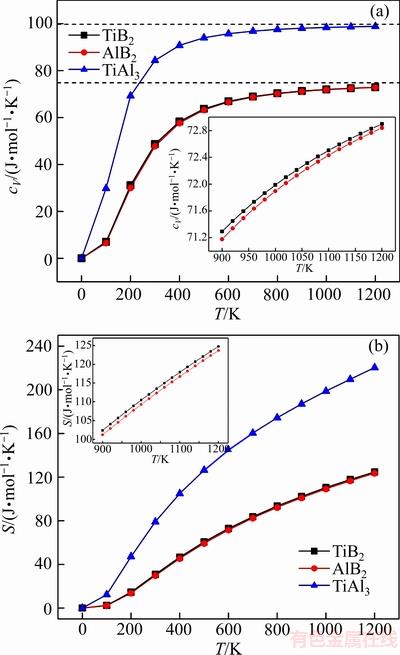
ͼ3 TiB2��AlB2��TiAl3������cV����S�Ĺ�ϵ
Fig. 3 Relationships of heat capacity(cV) and entropy(S) of TiB2, AlB2 and TiAl3 with temperature
ͼ3��ʾΪ��ѹ�����¼�������TiB2��AlB2��TiAl3�Ķ���Ħ������cV����S��0~1200 K���¶ȵı仯��ϵ��ͼ3(a)��ʾ������(300 K)������3�������cV�ֱ�Ϊ48.715��47.878��84.394 J/(mol��K)�������¶����ߣ�3����������ݾ��в�ͬ�̶ȵ�����0~300 K�ĵ��������£�������Ҫ�ܵ�����ԭ������Ӱ�죬���ڴ˽�3�����cV�����¶ȵ����߶�Ѹ�������ӵ°�T3���ɣ����¶�����300 K����ʱ���������ٿ�ʼ��С����TiAl3������������Ȼ��������������������������ڸ����µ������ͬ����T��1000 K�ĸ��������ڣ�3�������ڲ��Ǽ�гЧӦ�����ƣ�cV�����Ż��������ڸ��Ե�Dulong-petit���ޡ���0~1200 K�¶ȷ�Χ�ڣ����������������ݱ仯�������һ�£���������������������ྦྷ����ͬ���ڲ�ԭ�ӵ���ϸ�����ƶ���ɵġ���һ�������������(900~1200 K)��TiB2��AlB2���������¶ȴ��³����Թ�ϵ����ǰ������ʼ�մ��ں��ߡ�
ͼ3(b)��ʾ����ѹ������TiB2��AlB2��TiAl3����S���¶ȵı仯��������Կ�����100~500 K�¶ȷ�Χ�ڣ�TiAl3����ֵ���¶ȵ����߶����������¶ȼ�������ʱ���������ʷŻ�(����Ԥ�Ƶ��¶ȸ���ij��ֵ���ػ�������һ������ֵ)����TiB2��AlB2����ֵ�������ʵ���TiAl3���ҵ�T��200 K���������ʱȽϺ㶨����900~1200 K�������ڱ仯���������TiB2����ֵ����AlB2���������������ֵ���¶Ȼ������������ӵĹ�ϵ��900 Kʱ��TiB2��AlB2��TiAl3���طֱ�Ϊ102.367��101.191��187.088 J/(mol��K)��
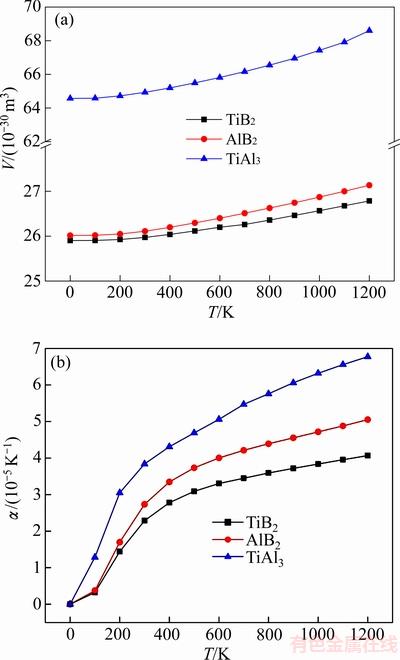
ͼ4 TiB2��AlB2��TiAl3����ƽ�����V��������ϵ�� ���¶ȵı仯
���¶ȵı仯
Fig. 4 Changes of thermal equilibrium volume and expansion coefficient of TiB2, AlB2 and TiAl3 with temperature
��ͬ�¶��¼�������TiB2��AlB2��TiAl3����ƽ�����V��������ϵ����ͼ4��ʾ����ͼ4(a)���Կ�����3���������ƽ��������¶ȵ����߶����������¶�T��300 K����ʱ��TiB2��AlB2�Լ�TiAl3��������������¶ȵ�Ӱ�졣��T��300 K������������¶ȴ��±������Թ�ϵ������800 K���ϵĸ�������TiAl3������������¶����߶���������ͼ4(b)��ʾ��0~1200 K�¶ȷ�Χ�ڣ�3�������������ϵ�����¶ȵ����Ӷ����ӡ����¶Ƚϵ�ʱ(200 K����)�����¶�����Ѹ�����������¶ȼ������ߣ�3������������Ż�����TiAl3��������Ȼ�������������Ҳ��������¶ȵı仯����õ���ӳ��������ϵ���������о���ʵ��Ӧ���е���Ҫ����ѧ����[33]�����Dz������ȶ��Ե���Ҫ����ָ�꣺ֵԽ������ϵ����ȶ���Խ���������ʾ3���������СΪ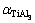 ��
��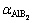 ��
�� ����ˣ�TiB2��AlB2��TiAl3�����ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3������ǰ�������ʾ���ߵĹ��ۼ��������Խ�������
����ˣ�TiB2��AlB2��TiAl3�����ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3������ǰ�������ʾ���ߵĹ��ۼ��������Խ�������
���÷��η��Ʊ�Al-Ti-Bʱ�����������Ϸ���(K2TiF6��KBF4)֮�䷢���ķ�Ӧ�Լ����յõ���������ɿ��Դ����ϼ��������Է�������������ʱ�����нϸ���������ֵ��TiAl3��Ҫһ���Ĺ��ȶȲ����γɣ���K2TiF6��������������������������ȷ�Ӧ���������ȣ�ʹ�������¶�Ѹ�����ߣ������ڴ�������TiAl3[34]�����ŷ�Ӧ���У������¶������ߣ��ʽ����������Ӽ���ϼ������Ե�TiAl3���ȶ��Խϲ�����۽�����Һ�У���ʹ��Һ��Ti��Ũ�����ߣ������н����д�����TiAl3��״С���壻��������ʱ��TiAl3�۽����ʼӿ죬����δ�����ܽ��TiAl3�ܵ���������TiԪ�صij�����ɢ��Ӱ��������������Ӷ������Ϊ��״����Ƭ״��
TiB2����������ֵ������AlB2������TiB2�������ʸ��ͣ���������Һ��Ӧ��������TiB2��ʣ���KBF4��Al����Ż�����AlB2��ͬʱ������������AlB2�����ȶ��Բ���TiB2���������ȶ�״̬������Ti�Ĵ��ڣ�Ti���û�AlB2�е�Alԭ�Ӷ����γ�(Alx, Ti1-x)B2������ֱ��TiB2[9]����ˣ��ڸ�w(Ti)/w(B)��Al-Ti-B�м�Ͻ���AlB2�ĺ���Զ����TiB2�����Ʊ���w(Ti)/w(B)��Al-Ti-B(��Al-1Ti-3B)ʱ������Ti������Խ��٣���Al���������ӹ����KBF4��ճ�������ھֲ�������Ti�ֲ������ȣ���ˣ�AlB2������ȫת��TiB2�����ڲ���AlB2�����̹����б���������
3 ����
1) �γ��������ܵļ�����������3�ֽ����仯������ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3��TiB2�Ľ���ܵ���AlB2��������Al-Ti-B�м�Ͻ���TiB2�����ȶ���
2) ���ӽṹ����������3���������������������ɴ�С����ΪTiAl3��AlB2��TiB2��TiAl3��AlB2�ļ������������Ӽ�Ϊ������TiB2����������B 2p��Ti 3d���̬�����ӻ��Ĺ��ۼ�Ϊ����
3) ��г�°�ģ�ͼ�����ʾ��TiB2��AlB2��TiAl3�Ķ���Ħ�������������¶����߶���ͬ�̶ȵ�����ͨ������������ϵ������3����������ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3��
REFERENCES
[1] AURADI V, KORI S A. Influence of reaction temperature and reaction time for the manufacturing of Al-Ti-B (Ti:B=5:1, 1:3) master alloys and their grain refining efficiency on Al-7Si alloys[J]. Transactions of the Indian Institute of Metals, 2012, 65(6): 637-645.
[2] BIROL Y. An improved practice to manufacture Al-Ti-B master alloys by reacting halide salts with molten aluminium[J]. Journal of Alloys and Compounds, 2006, 420(1/2): 71-76.
[3] JONES G P, PEARSON J. Factors affecting the grain-refinement of aluminum using titanium and boron additives[J]. Metallurgical and Materials Transactions B, 1976, 7(2): 223-234.
[4] �� ��, ������, ���ڷ�, ������. �������巴Ӧ���Ʊ�Al-5Ti-1Bϸ����[J]. ���Ϲ���, 2017, 45(2): 39-45.
LI He, CHAI Li-hua, MA Teng-fei, CHEN Zi-yong. Synthesis of Al-5Ti-1B refiner by melt reaction method[J]. Journal of Materials Engineering, 2017, 45(2): 39-45.
[5] KORI S A, MURTY B S, CHAKRABORTY M. Development of an efficient grain refiner for Al-7Si alloy and its modification with strontium[J]. Materials Science and Engineering A, 2000, 280(1): 58-61.
[6] YU L, LIU X. The relationship between viscosity and refinement efficiency of pure aluminum by Al-Ti-B refiner[J]. Journal of Alloys and Compounds, 2006, 425(1/2): 245-250.
[7] Ф����, ������, �ƽ���, �� ��, ������. Al-Ti-C��Al-Ti-B����ϸ������Zr�ж�����[J]. �й���ɫ����ѧ��, 2012, 22(2): 371-378.
XIAO Zheng-bing, DENG Yun-lai, TANG Jian-guo, CHEN Qi, ZHANG Xin-ming. Poisoning mechanism of Zr on grain refiner of Al-Ti-C and Al-Ti-B[J]. The Chinese Journal of Nonferrous Metals, 2012, 22(2): 371-378.
[8] �γ�ΰ, ������, �� ��, ����ƽ, �˴���. ԭ������˳��Է��η��Ʊ�Al-Ti-B�м�Ͻ��Ӱ��[J]. �й���ɫ����ѧ��, 2016, 26(1): 204-211.
LIAO Cheng-wei, CHEN Wen-tian, CHEN Huan, FU Chun-ping, PAN Chun-xu. Effect of feeding order on preparation of Al-Ti-B master alloy by fluoride salt method[J]. The Chinese Journal of Nonferrous Metals, 2016, 26(1): 204-211.
[9] FJELLSTEDT J, JARFORS A. On the precipitation of TiB2 in aluminum melts from the reaction with KBF4 and K2TiF6[J]. Materials Science and Engineering A, 2005, 413(6): 527-532.
[10] MIRKOVIC D, GROEBNER J, SCHMID-FETZER R, FABRICHNAYA O, LUKAS H L. Experimental study and thermodynamic re-assessment of the Al-B system[J]. Journal of Alloys and Compounds, 2004, 384(1/2): 168-174.
[11] HERRING C. A new method for calculating wave functions in crystals[J]. Physical Review, 1940, 57(12): 1169-1177.
[12] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review, 1965, 140(4A): A1133/A1138.
[13] XIE Y P, WANG Z Y, HOU Z F. The phase stability and elastic properties of MgZn2 and Mg4Zn7 in Mg-Zn alloys[J]. Scripta Materialia, 2013, 68(7): 495-498.
[14] PERDEW J P, BURKE K, ERNZERHOF M. ERRATA: Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-3868.
[15] MONKHORST H J. Special points for Brillouin-zone integrations[J]. Physical Review B Condensed Matter, 1976, 16(4): 1748-1749.
[16] BLANCO M A, FRANCISCO E,  V. GIBBS: Isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model[J]. Computer Physics Communications, 2004, 158(1): 57-72.
V. GIBBS: Isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model[J]. Computer Physics Communications, 2004, 158(1): 57-72.
[17]  M, RECIO J M, FRANCISCO E, BLANCO M A,
M, RECIO J M, FRANCISCO E, BLANCO M A,  A M. First-principles study of the rocksalt-cesium chloride relative phase stability in alkali halides[J]. Physical Review B, 2002, 66(14): 144112-144123.
A M. First-principles study of the rocksalt-cesium chloride relative phase stability in alkali halides[J]. Physical Review B, 2002, 66(14): 144112-144123.
[18] WANG M. The investigation of dependences of mechanical and electronic properties of TiB2 on pressure using the first-principles method[J]. Physica Scripta, 2014, 89(11): 115702-115714.
[19] DUAN Y H, SUN Y, GUO Z Z, PENG M J, ZHU P X, HE J H. Elastic constants of AlB2-type compounds from first-principles calculations[J]. Computational Materials Science, 2012, 51(1): 112-116.
[20] DUAN Y H, SUN Y, LU L. Thermodynamic properties and thermal conductivities of TiAl3-type intermetallics in Al-Pt-Ti system[J]. Computational Materials Science, 2013, 68(2): 229-233.
[21] MEDVEDEVA N I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV O N, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl[J]. Acta Materialia, 1998, 46(10): 3433-3442.
[22] WU M M, JIANG Y, WANG J W, WU J, TANG B Y, PENG L M, DING W J. Structural, elastic and electronic properties of Mg(Cu1-xZnx)2 alloys calculated by first-principles[J]. Journal of Alloys and Compounds, 2011, 509(6): 2885-2890.
[23] FJELLSTEDT J, JARFORS A E W, SVENDSEN L. Experimental analysis of the intermediary phases AlB2, AlB12 and TiB2 in the Al-B and Al-Ti-B systems[J]. Journal of Alloys & Compounds, 1999, 283(1/2): 192-197.
[24] KELLY P M, REN H P, QIU D, ZHANG M X. Identifying close-packed planes in complex crystal structures[J]. Acta Materialia, 2010, 58(8): 3091-3095.
[25] VAJEESTON P, RAVINDRAN P, RAVI C, ASOKAMANI R. Electronic structure, bonding, and ground-state properties of AlB2-type transition-metal diborides[J]. Physical Review B, 2001, 63(4): 33-41.
[26] BURKHARDT U, GURIN V, HAARMANN F, BORRMANN H, SCHNELLE W, YARESKO A, GRIN Y. On the electronic and structural properties of aluminum diboride Al0.9B2[J]. Journal of Solid State Chemistry, 2004, 177(2): 389-394.
[27] ZHANG M X, KELLY P M, EASTON M A, TAYLOR J A. Crystallographic study of grain refinement in aluminum alloys using the edge-to-edge matching model[J]. Acta Materialia, 2005, 53(5): 1427-1438.
[28] ZHANG H L, HAN Y F, WANG J, DAI Y B, SUN B D. An Ab initio molecular dynamics study on the structural and electronic properties of AlB2, TiB2 and (Alx,Ti(1-x))B2 in Al-Ti-B master alloys[J]. Journal of Alloys and Compounds, 2014, 585(3): 529-534.
[29] LI Y, GAO Y, XIAO B, MIN T, FAN Z, MA S, XU L. Theoretical study on the stability, elasticity, hardness and electronic structures of W-C binary compounds[J]. Journal of Alloys and Compounds, 2010, 502(1): 28-37.
[30] SPOOR P S, MAYNARD J D, PAN M J, GREEN D J, HELLMANN J R, TANAKA T. Elastic constants and crystal anisotropy of titanium diboride[J]. Applied Physics Letters, 1997, 70(15): 1959-1961.
[31] LOA I, KUNC K, SYASSEN K, BOUVIER P. Crystal structure and lattice dynamics of AlB2 under pressure and implications for MgB2[J]. Physical Review B, 2002, 66(13): 134101.
[32] GHOSH G, WALLE A V D, ASTA M. First-principles phase stability calculations of pseudobinary alloys of (Al,Zn)3Ti with L12, D022, and D023 structures[J]. Journal of Phase Equilibria and Diffusion, 2007, 28(1): 9-22.
[33] OKADA Y, TOKUMARU Y. Precise determination of lattice parameter and thermal expansion coefficient of silicon between 300 and 1500 K[J]. Journal of Applied Physics, 1984, 56(2): 314-320.
[34] EL-MAHALLAWY N, TAHA M A, JARFORS A E W, FREDRIKSSON H. On the reaction between aluminium, K2TiF6 and KBF4[J]. Journal of Alloys and Compounds, 1999, 292(1/2): 221-229.
First principle study of AlB2, TiB2 and TiAl3 in Al-Ti-B alloy
HUANG Yuan-chun1, 2, 3, SHAO Hong-bang1, XIAO Zheng-bing1, 2, 3, REN Xian-wei1, GUO Xiao-fang1
(1. Light Alloy Research Institute, Central South University, Changsha 410083, China;
2. State Key Laboratory of High Performance and Complex Manufacturing, Central South University, Changsha 410083, China;
3. College of Mechanical and Electrical Engineering, Central South University, Changsha 410083, China)
Abstract: The crystal structure, heat of formation, cohesive energy and electronic properties of AlB2, TiB2 and TiAl3 in Al-Ti-B system under 0 K condition were investigated by using first principles based on density functional theory, and the quasi-harmonic Debye model was performed to calculate the thermodynamic properties of these phases. The results show that the heat of formation and cohesive energy of AlB2, TiB2 and TiAl3 are both negative, which indicate that these phases possess structural stability, and the structural stability decreases in the order from big to little of TiB2, TiAl3, AlB2. The iconicity and metallicity of these compounds increase in the following sequence from big to little of TiAl3, AlB2, TiB2, and the dominant bonding in TiAl3 and AlB2 is ionic, while the bonding in TiB2 is mainly covalent. The thermodynamic calculation results show that the thermal stabilities of the three phases decrease in the order from big to little of TiB2, AlB2, TiAl3, which can be used to illustrate the reactions between Al melt and fluoride salts when preparing Al-Ti-B by fluoride salt process.
Key words: Al-Ti-B alloy; first-principles; quasi-harmonic Debye model; structural properties; thermodynamics properties
Foundation item: Project(2015CB057305) supported by the National Basic Research Development Program of China
Received date: 2017-06-05; Accepted date: 2017-09-20
Corresponding author: HUANG Yuan-chun; Tel: +86-13507315123; E-mail: science@csu.edu.cn
(�༭ ����)
������Ŀ�������ص�����о���չ�ƻ�������Ŀ(2015CB057305)
�ո����ڣ�2017-06-05�������ڣ�2017-09-20
ͨ�����ߣ���Ԫ�������ڣ���ʿ���绰��13507315123��E-mail: science@csu.edu.cn
ժ Ҫ�����û����ܶȷ������۵ĵ�һ��ԭ���������о�0 K������Al-Ti-B��ϵ��AlB2��TiB2��TiAl3��ƽ�⾧�������γ��ȡ�����ܼ��������ԣ��������г�°�ģ�Ͷ�3�������ڸ��������µ�����ѧ�������ʽ����˷�����������������AlB2��TiB2��TiAl3���γ��������ܾ�Ϊ��ֵ������3��������нṹ�ȶ��ԣ����ȶ����ɴ�С����ΪTiB2��TiAl3��AlB2��3���������������������ɴ�С����ΪTiAl3��AlB2��TiB2��TiAl3��AlB2��ʾ���Ӽ����ԣ���TiB2���������Թ��ۼ�Ϊ����3����������ȶ����ɴ�С����ΪTiB2��AlB2��TiAl3����������TiB2��AlB2��TiAl3������ѧ���ʿ������ڷ������η��Ʊ�Al-Ti-BʱAl���������֮�䷴Ӧ��������Լ��м�Ͻ����յ�������ɡ�
