
Motif Finding����ķֲ�ʽ�����㷨
����ƽ���� ��
(���ϴ�ѧ ��Ϣ��ѧ�빤��ѧԺ������ ��ɳ��410083)
ժ Ҫ��
ժ Ҫ�����ڴ�DNA�����γ�k��ͼ��ͼ�����㷨�Ͳ���k-clique�������㷨�������ʵ���˶�Motif Finding�������ķֲ�ʽ�����㷨�����㷨����Ҫ�ص��ǣ������µ�1-3����֦�㷨��ʵ�ֲַ�ʽ������ƣ�������������ż�����̵�չ����ÿһ�β��ϵطֽⲢ�ֲ�����ȷ������������������Ŀͻ����ϣ��������˸�����������������ϡ�ʵ�����������ֲ�ʽ�����㷨������ö�̨����Эͬ���㣬�ܹ���ȷ����Ч�صõ���������Ϊ�������������ѽ��Motif Finding�����ṩ����Ч�Ľ���ֶΡ�
�ؼ��ʣ�
Motif��k��ͼ����������������ֲ�ʽϵͳ��
��ͼ����ţ�TP311 ���ױ�ʶ�룺A ���±�ţ�1672-7207(2007)05-0943-07
Distributed parameterized algorithm for Motif Finding
ZHANG Zu-ping, WANG Li
(School of Information Science and Engineering, Central South University, Changsha 410083, China)
Abstract: Based on the graph-theoretic algorithm, which is used to generate k-partite graph from DNA sequences, and a theoretic algorithm which is used to search k-clique in k-partite graph, a new distributed parameterized algorithm is designed and implemented to solve Motif Finding problem. The main characteristics of this algorithm are the introduction of 1-3 tree branching algorithm and the implementation of distributed mechanism, i.e., task can be constantly decomposed into several tasks as program proceeding at each phase, and then these tasks are distributed to several client computers which is applied for participation of calculation. On the server, the tasks are distributed and balanced as well as the results are received and combined. Experimental results show that this distributed parameterized algorithm is correct and efficient since it makes full use of computing capability of many computers, and provides an effective approach to solving the difficult problems in biocomputing such as Motif Finding.
Key words: Motif; k-partite graph; clique; biocomputing; distributed system
������Ϣѧ�����˵Ľ���ѧ�ƣ����ۺ�������ѧ���������ѧ������ѧ�ĸ��ֹ��ߣ��������������������������������ѧ����[1]�����У��Ի���ͻ���ȵ����ļ��[2]�ǵ�ǰ�о����ȵ㣬������չ�Ķ������бȶԷ��滹��������̽���Ρ��Ѿ�֤��Motif Finding��NP������[3]��������������н��类�����о�������֮һ��Ҳ�ǽ������о����ȵ�����[4-6]����Ȼ�Ѿ�����һЩ������������ߣ��磺����̰���㷨��CONSENSUS[7]������Gibbsȡ����GibbsDNA[8]�ͻ���EM�㷨��MEME[9]�ȣ����ǣ���Щ�㷨������ȷ��һ�����ҵ�ȷʵ���ڵ�Motif����Ϊ���Ƕ���Ҫһ���ܺõĿ�ʼ�㣬���ԣ����Dz�ȷ���㷨�����⣬�����꾡���о����п��ܵ�Motifs�㷨����Ȼ�ܱ�֤�ҵ����ŵ�Motif�����ǣ�����ʱ������Motif����l�����Ӷ���ָ����������l�ϴ����ս�����б�ò����С�Ϊ����������⣬��������һ�������������㷨���Motif Finding��������о�ȷ�⣬��Ч�����ȷ���㷨��©��������⣻��һ���棬�����ֲ�ʽ���㷽����ֲ�ʽ���ϵͳ[10]�����������ֳɽ�С�Ŀ��Զ���ִ�еĵ�Ԫ�����ֲ����������п��м�����Դ�Ķ�̨������ȥ��ϵͳ�����������صļ��������кϲ��������γ���������ս�����Ӷ��ӿ���Motif Finding����������̡�
1 �ؼ��㷨��������
1.1 �������
����Pevzner ��(l, d)-motifģ��[11]�������k�����Ⱦ�Ϊn��������У���ÿ�������������ѡ1��λ������1��(l, d)-motif����Motif����1����Ϊl������d��ͻ���ģʽP������Motif Finding����Ҫ���ڲ�֪��ģʽP������λ�õ�������ҳ���Щ�����Motif��
��������[12]���㷨��Ϊ2������1����Ӧ��Pevzner��ͼ�����㷨[11]��DNA���в���k��ͼ����ÿ������Si�еĵ�j��λ�ã�����1������Sij����ʾ1����λ��j(1��j��n-l+1)��ʼ�ij�Ϊl���Ӵ�����i��p����Sij��Spq֮��ľ��벻����2d(ͻ��Ԫ�ص�����)������1���߽�2������Sij��Spq��������2�����ο�����k-clique�������㷨[12]��Ӧ�÷��η�����k��ͼ�����k-clique�����õ���k��ͼϸ��Ϊk��(k�䣼k)��ͼֱ��k��С�������Ϊֹ�����k��-clique֮���ٺϲ�Ϊ�����k-clique����õ�k-clique��Ϊ�����Motif��������λ�á�
1.2 k��ͼ�γ��㷨
1.2.1 �㷨˼��
����ͼ���ۣ��㷨�γ�k��ͼ�Ĺؼ������ж�2��������Ƿ���ڱߣ�������2���ַ���֮��IJ�ƥ������Ƿ���������Χ֮�ڣ���ͼ�����㷨��δ������Ч�ķ�����ͨ���о��������㷨��PROJECTION[13]��WINNOWER��SP-score[11]�����µ��о������㷨[14]�ȶ�ֻ�����˼��滻ͻ�䣬���ܴ������ڲ����ɾ��ͻ������С�Ϊ���������⣬���Ĵ�Mismatch Tree[15]�㷨�еõ������������1��3����֦�㷨����Ϊ�����㷨��������һ��������ʽ�����У���1������ƥ��ʱֻ����1����֦����3��������ƥ��ʱ����3����֦��
��2���ַ���A��B���бȽ�ʱ������ijһλ�ó����˲�ƥ�䣬���λ���ϵ��ַ����ܲ������滻�������ɾ��ͻ�䡣���ȣ���3��ͻ������㷨����
a. �滻������A��B���κ�1���������滻ͻ�䣬������������ͻ�䣬����۶�������A��B�ڸ�λ���ϵ��ַ���������ƥ���������������1��
b. �����ɾ������2���ַ������бȽϣ������ɾ��ͻ�䶼���������һ���ַ������Եġ�����ij��λ����A���B�����˲���ͻ�䣬���൱��B���A������ɾ��ͻ�䣬��ʱ��Ӧ�ú��Ը�λ����A���ַ�����A��һ��λ���ϵ��ַ�������B�ĸ�λ�����ַ��Ƚϣ�ͬʱ��ƥ�������������1��
1-3����֦�㷨Ϊ��
boolean done=false; /* ������־ */
int mis=0; /* ��ƥ����� */
int pos1=0; /*�ַ���A��Ҫ���бȽϵ��ַ�λ��*/
int pos2=0; /*�ַ���B��Ҫ���бȽ��ַ���λ��*/
while (true) {
if (done) then return;
if (pos1�����ַ���A�ľ�ͷ or pos2�����ַ���B�ľ�ͷ or mis>�����������ƥ�����) then break;
if (pos1λ���ϵ��ַ���pos2�ϵ��ַ�һ��) then {
pos1++;
pos2++;
continue;
}
else {
mis++;
���������Dz���ͻ����̣߳���ֵpos1+1, pos2, mis;
����������ɾ��ͻ����̣߳���ֵpos1, pos2+1, mis;
�������滻ͻ�䣬pos1++, pos2++;
continue;
}
}
if (mis <= ���������ƥ�����) then{
if (done) then return;
synchronize {
if (done) then return;
done = true;
����������k��ͼ;
}
}else return;
Ϊ�˸��õ���������㷨����������Ƚ��ַ���actta���� ��attag����ʱ����������߳���������a, c, t, gΪ4���������ͼ1��ʾ��
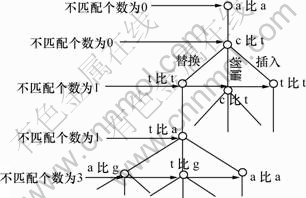
ͼ1 Ӧ��1-3����֦�㷨�Ƚ�2���ַ���ʾ��
Fig.1 Comparison between two sequences with 1-3 tree branching algorithm
�߳�����һ�����ϵĸ��ʵ���ϲ������ڣ����Կ������߳����еĹ켣��1����֦����1���̣߳�ÿ���̶߳�ӵ���Լ���һЩ���ϣ��粻ƥ���������������2���ַ������ԱȽϵ����ĸ��ַ��ȡ�
1.2.2 �㷨����
�Ƚ�2����Ϊl���ַ�����������ƥ�����Ϊ2d�����㷨��ʱ�临�Ӷ�����������Ϊl��������Ϊ![]() ��dһ�㲻����һ����Ϊn�������У������г�Ϊl���ַ�������k��ͼ�е�1�����㣬����
��dһ�㲻����һ����Ϊn�������У������г�Ϊl���ַ�������k��ͼ�е�1�����㣬����![]() �����㡣
�����㡣
��1�������еĶ���������k-1�������е����ж�����бȽϣ�������(k-1)(n-l+1)2�ζ���Ƚϣ�
��2�������еĶ���������k-2�������е����ж�����бȽϣ�������(k-2)(n-l+1)2�ζ���Ƚϣ�
����
��������2�����м���k-1�������еĶ�����������������е����ж�����бȽϣ�������(n-l+1)2�ζ���Ƚϣ�
����������м���k�����в������κ����н��бȽϡ�
���ԣ��Ƚϴ�����Ϊ��
![]()
![]() =
=![]()
![]() =
=![]() ��
��
������γ�k��ͼ���������µ�ʱ�临�Ӷ�Ϊ��O(lk2(n-l)2)�������µ�ʱ�临�Ӷ�Ϊ�� O(32d k2(n-l)2)��
���ڸ��㷨����ʱ�������̲߳�ȷ������ú������ķ������ʾ�����0����ʵ����֤����k, n��d����(k = 10, n = 82, d = 4)��Ҫ��ȷ�������£�ʵ�ʼ�����С�
1.3 k-cliqueѰ���㷨
1.3.1 �㷨˼��
����k-clique�������㷨�����˴������µĵݹ鷽����ʵ�ַ��β��ԣ�ʵ���ϸ�Ϊ��Ч�ķ�ʽ�Dz������¶��ϵ�ѭ��������ʵ�ַ��Σ��������Ի�ø��ߵ�Ч�ʣ����ڽ��㷨���зֲ�ʽ����������ֻ�轫�㷨�����ı�������Ӹ��ӵ����������[12]�����巽�����¡�
��k��ͼ����ΪһЩk���ͼ(k�䣼k)����k���ͼ�����k��-clique���ٽ�k��-clique��ϳ�k��-clique(k�䣼k�壼k)��ֱ�������ϳ�k-clique�����������k��-clique��ϳ�k-clique��������ж�k��ͼ���ٴ��ڴ�СΪk������clique���ij���������Щ�м������Է�������k��ͼ������š�k���ѡ��Ӧ��ʹ����ͼ�㹻ϡ�������ں�������������ܴ洢�ṹ����Լ(�洢���DZ�)��k��ѡ��4��ǡ����
�����и�������4�ı���ʱ����ֻʣ1�����У�����������ж��㿴��1-clique��������2�����У������2�����м�ı߿���2-clique��������3�����У����2�����м��2-clique��1�����е�1-clique��ϵõ�3-clique��
1.3.2 �㷨����
��kΪ2������(��1)����ʱ��ʱ�临�Ӷ���������������ʱ�临�Ӷ������������������������k��ͼ����ȫͼ������ζ��ÿ2������䶼�бߣ�ÿ��������![]() �����㣬��ÿ2�����м���(n-l+1)2���ߣ�ÿ4����������(n-l+1)4��4-clique��
�����㣬��ÿ2�����м���(n-l+1)2���ߣ�ÿ4����������(n-l+1)4��4-clique��
�㷨������2�����м�����б�(��2-clique)����2�����м�����б߽��бȽϣ��õ�һ��4-clique��Ȼ����һ��4-clique����һ��4-clique���бȽϣ��õ�һ��8-clique��������ֱ�����Ƚ�2��k/2-clique������Ƿ����k-clique��
��1�����������4-clique��ʱ�临�Ӷ�Ϊ��
![]() ��
��
��2�����������8-clique��ʱ�临�Ӷ�Ϊ��
![]() ��
��
��
���1������![]() �����������k-clique��ʱ�临�Ӷ�Ϊ��
�����������k-clique��ʱ�临�Ӷ�Ϊ��
![]() ��
��
�Ƴ�ͨ�ù�ʽΪ(m������m��)��ʱ�临�Ӷ�Ϊ��
![]() =
=
![]() ��
��
�ܵ�ʱ�临�Ӷ�Ϊ��
![]()
![]()
![]()
![]() ��
��
ʵ�ʼ����ʱ�临�Ӷ�ԶС�ڴ�ʱ�临�Ӷȣ�����������Ӧ�÷ֲ�ʽϵͳ������⣬������ʱ���ڿɳ��ܷ�Χ֮�ڡ�
2 �ؼ����ݽṹ���
2.1 k��ͼ
k��ͼ����![]() �����㣬Ϊ�����ȡ������Ϣ���ڴ洢�ļ���ͷ���洢1������������
�����㣬Ϊ�����ȡ������Ϣ���ڴ洢�ļ���ͷ���洢1������������
k��ͼ�����ݽṹ�����ڽӱ�������1�������д洢��1�������λ������ʾ1���ߣ�����Ž�С�Ķ����д洢������֮��������Žϴ�Ķ���λ�ã����ڶ���Sij��洢����(p, q)������i��p��������Sij��Spq���бߣ����һ�������㰴�������ĸ����н��з��ֻ࣬�洢�ڴ������е�λ�ã���ֻ�洢q����ʡ��һ��ռ䡣
�������Vector���ݽṹ�������������(0, 3)�Ĵ洢�ṹʾ��ͼ������ö�����(1, 2)��(1, 5)��(1, 6)��(2, 4)��(3, 8)��(3, 9)�ȶ�����������ö�������ݽṹ��ͼ2��ʾ��
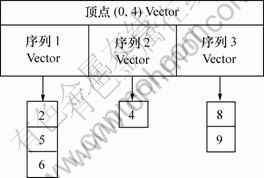
ͼ2 ����Vector�����ݽṹ
Fig.2 Data structure of vertex
2.2 ��
�����ŵĴ�С�����и�����һ���ģ���ˣ���1��ȷ�����ȵ��������洢�����е�Ԫ���Ƕ��㣬�ó���Ϊ2�������ʾ�������������ŵ����ݽṹ��1����ά���飬��1ά����Ϊ��������и�������2ά����Ϊ2��������ŵĸ�����ȷ������ˣ���Vector���洢��Щ�š�
3 �㷨�ֲ�ʽ˼�뼰��ʵ��
�����㷨��Ȼ�ܹ�������Ч��⾫ȷ�⣬���ǣ�ʱ�临�Ӷ���Ȼ�������ģ��ָ����������ˣ�Ϊ�ӿ�����ٶȣ�����ֲ�ʽ���ƣ����㷨���зֲ�ʽ������ʹ֮���ڷֲ�ʽƽ̨�����У�����ƶ�̬����ģ�ͣ���֤���ؾ��⡣
�ֲ�ʽϵͳ����һ�����������ƶ�̨�ͻ�����ģʽ�����з�����ֻ���������������տͻ������ؽ�������ϣ��ͻ����Էָ��Լ���������м��㡣�ͻ����˵ij���˼����ǰ�����������������ɴӷ������õ������������������ģ���ܵ�����С�öࡣ����������ܵķֲ�ʽ�����㷨����ָ�������˳�����õ��㷨��
3.1 ����k��ͼ
Ϊ��ʹ�ֽ���������ģ��������ö����н��д�ֱƽ�ֵķ�������ͼ3��ʾ������ˮƽ�Ĵ���������1��DNA���У�ǰ�˵�ʵ�߲��ִ���Ҫ�ȽϵĶ��㣬��Ϊ(n-l+1)��Ϊ��ƽ���ָ��������ォʵ�߲����ô�ֱ�����߽���ƽ�֣����ֳ���ÿ1�ض�Ӧ�ڱȽ�ʱ��һ�����㡣

ͼ3 ��ֱƽ��DNA������Ķ���Ƚ�����
Fig.3 Vertically average division of comparing task
��n-l+1���ܱ����ַ�������ʱ��������ƽ���ָ�ǰ��������֤�������������С����������1�������Ȳ����ȱ�ȡ��ִ�У������˶�ϵͳЧ�ʵ�Ӱ�졣
���ֽ������������ͻ������м��㡣��ͼ3��ʾ����Ϊ��������1̨�ͻ����ֵ��˵�2��������ô�������õ�1�����������߷ֳɵĵ�2�����ֵĶ������2��3��4�����е�ȫ��������бȽϣ�Ȼ���õ�2�����еĵ�2���ֶ������3��4�����е�ȫ��������бȽϣ�����õ�3�����еĵ�2���ֶ������4�����е�ȫ��������бȽϡ��Ƚ����ѽ�����ط������������������������ͻ������ؽ���������ϣ��õ�k��ͼ��
���ϲ����ǵ��������õ�һ�����ؽ��ʱ���ѽ���ļ��г�����������IJ���ȫ������(Ϊ��ֹ�ڴ�������������ο�������ļ�������һ���ֲ���)��һ��ָ�������ļ�����ý���ļ������������ļ�����������������������ļ��ڵ�1���������ʱ�����������ڿ�ʼ���ַ���һ���յ�long[k-1][n-l+1]������������ȫ�����㡣
3.2 ����k-clique
����һ�����ηֽ�����ʹ����������Ⱥ����Ǻܹؼ��ġ���Ϊ���������������ȹ�С����ʹϵͳƵ���������Ӵ����ļ�������Ӱ��ϵͳЧ�ʣ������ȹ�������������÷ֲ�ʽϵͳ����Դ�����⣬��k��-clique�Ľ�Ϸ�����Ӱ��ϵͳ���ؾ���ͼ�����Դ�����á��������һ����̬�ľ���ģ���Խ��������⡣
Ϊ�˳�����÷ֲ�ʽϵͳ��Դ����Ӧ�÷��η������зֳ�4��1���ͬʱ������ÿ���������ϸ�֡������������¡�
��ÿ��ĵ�1��������Ϊ�����У��ӵ�1�����㿪ʼ������������������йصļ����������һ��һֱ�����1�����㣬�����γ�n-l+1�������йص�������ָ�����������������ж��йصĶ���ͱߡ�������˵����1�������ǵ�1�����еĵ�1������͵�2�����ж��������б����3�͵�4�����м����бߵ�����ж���ֱ����n-l+1������Ϊֹ�������ԣ������������Ǻ��ʵģ�����1�ֺ��ʵļ��������ȵ��㷨��������Ч�����ϵͳЧ�ʡ�
���ڵõ�k��-clique����Ҫ�ٽ�k��-clique��ϳ�k��-clique(k�䣼k�壼k��������Ҫ�����2��k��-clique�õ�k��-clique��Ϊ�µ��ж���������ͻ�����һ�����㡣���ֻ�Ǽط�����2����ͽ����Ͻ�����һ��ѭ������ô���ض�ʹ����һ��ĺ�һ���ǰһ�����ܶ���ܲ������������Ʊ�Ӱ�����һ��Ľ�ϣ�����ʹϵͳЧ���½���Ϊ�˿˷����ʱ��ȱ�ݣ������һ����̬������ģ�ͣ�Ϊ�˱������⣬�ڽ������ģ��֮ǰ�Ƚ��ܿ˷�����ȱ�ݵľ�̬ģ�͡���ͼ4��ʾ��������������������鲢�Ҹ���������������ǰ�����ȥ��˳�����η��صģ�������б������2�����δ���ͬһ���飬������ǵ�1�������1����ɵ�(����ƴ��ϵ��µ�һ���ž���Ϊ��1�㡢��2�㣬����)�����ֱȵ�2������������ȷ��أ���ô�����齫�Ƶ���2�����ǰ�����������ϡ����ģ���ܹ��˷�ʱ��ȱ�ݵ�ԭ���ǣ�ÿ������ɵ�����ѡ��������н�ϵ���ʱ��ѡ�����û�н�Ϲ����������˳����е��Ǹ���
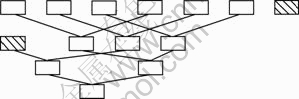
ͼ4 �˷���ʱ��ȱ�ݵľ�̬ģ��
Fig.4 Static combining model without time flaw
ÿ����ֳ�n-l+1��С�����ܷ���0~ n-l+1���������Ϊ0�������������У�ҪѰ�ҵ�k-clique�����ڣ���Ϊn-l+1��ֱ�ӽ���Щ���������һ�������н�ϣ�����������(n-l+1)2�����������������ȥ��������������ָ�����������뽫һ������Ľ���Ƚ��кϲ���Ȼ��������һ����������ϣ���������Ϻ���������ֻ��n-l+1���������⣬���ص�2���������ȫ�����زſ�ʼ��ϣ�������һ���������ȫ�����غϲ���Ϳ�������һ��ֻ�в��ַ��ؽ��������н�ϡ�
��ʵ�ֶ�̬ģ��ʱ�������ļ������洢����ķ��ؽ�����ļ��п��Զ�̬�������ͷš�������һ�������������˵�������̬ģ�͵Ĺ������̡���ͼ5��ʾ����1������Ľ��ȫ�����ز��Һϲ�Ϊ1��ʱ���ú�������������ʾ�����˳���Ƿ���У���ʣ������(���Ѿ���������û�н�����ص�����)�Ķ���������������һ������7�飬ǰ6�鶼��4������1�飬�ֽ��С����֮��ַ���ȥ�����1��ֻ��2�����У�ֱ�Ӵ����2��������õ������б���Ϊ2-clique�����6���ļ��У���ʱ��6���ļ�����������ȥѰ���ܹ��������н�ϵ��ļ��С����ǣ�û��һ���ļ������з��صĽ�������ԣ�����дע����Ϣ�����ڽ�����ij���ļ��з�������������ϡ�����0���ļ��еĵ�1���������ʱϵͳɨ��ע����Ϣ�����ֵ�6���ļ��в���֮��ϣ���̬�����ļ���7����Ž����ķ��ؽ�������赱��1���ļ�����ʱ��2��3��4���ļ��зֱ���3��2��5������û�з��أ�����5���ļ��л��ǿյģ����1���ļ��о�ѡ���ļ���2���н�ϣ������ļ���8�����ļ���3��ʱ���ļ���5��ʣ��������С���ļ���4��ʣ������������ѡ�����ļ���5��ϣ������ļ���9�����ļ���4��ʱ���������DZ�������һ�������뱾����ļ��н�ϣ���ˣ������IJ�����1������������һ������һ����ļ��н�ϡ�����ͼ5�������ļ���8��ϣ������ļ���10�����ļ���7���ļ���9���ʱ�������ļ���7�Ѿ�������������ļ���0��6���ѱ��ͷţ����ԣ��ظ������ļ���0����Ž����Ľ��������ļ���10��0��ϣ��ظ������ļ���1�������ս��������ļ���1�С�
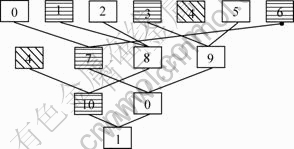
ͼ5 ��̬�����ģ��
Fig.5 Dynamic combining model
4 ʵ������֤������
4.1 �ж�2��������Ƿ��б�
3��������ƥ��IJ��Խ�����1��ʾ�����У�����ָ�༭���롣ƥ���������д�д�ַ�����ƥ����ַ���Сд�ַ�������ƥ����ַ��������ɾ��ͻ������Ӧ��-����
��1 ������ƥ�����֤ʵ��
Table 1 Test results of comparing sequences

����֤1������1-3����֦�㷨����ֻ�����滻ͻ������бȶԿ���ʤ�Σ���֤2�����������ڲ����ɾ��ͻ��ʱ��1-3����֦�㷨ͬ����Ч����֤3�������ڴ���1�����в����˼��������IJ���ͻ�������ļ������ʱ��1-3����֦�㷨��Ȼ��Ч����Mismatch Tree[15]�㷨������Ч�������������
4.2 Ѱ��k-clique
���Ի�����k=10, n=82��ģ�ⷶ��������Ѱ��(15, 4)-motif�����ܽ����ͼ6��ʾ��
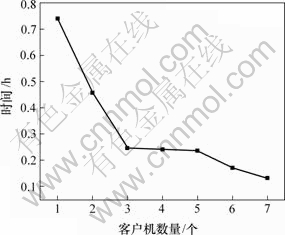
ͼ6 ��̨�ͻ�������ʱ����Խ��
Fig.6 Test results of multiple clients
�Ӳ������ܽ�����Կ������ֲ�ʽ������ƴ�������˳�������ʱ�䡣��Ȼ���ĵ��㷨Ҫ���1����k��ͼ������ȫ����ɷ��غ���ܿ�ʼ��2���ļ��㣬���ǣ��ڸò����У������ڵ�1���ֽ�����ʱ�м�̨�ͻ�����������ƽ���ֽ�ɼ�������������С����������1����������ȱ�ȡ���㣬���ԣ��յȴ�ʱ����١�
������������н���ʵ�ʼ���õ�3 000����ţ���ʵ���Ϸ�����������ֻ��2��������������֤��ȷ�����ĵ��㷨���������ȷ�ģ����������Ľ������ΪMotif Finding���Ȿ������һ����������ڶ�����⣬�þ�ȷ�㷨�õ������Ҫ������ѧ֪ʶ��һ��ѡ���������ҳ�������ȷ����Ч�Ľ����
5 �� ��
a. �о���Motif Finding����ľ�ȷ�㷨�������1-3����֦�㷨����Ч�ؽ����������ͼת��ʱ����ͻ��ʱ���Ա���Ĺؼ����⡣
b. �����Motif Finding����ķֲ�ʽ�����㷨�����ö����д�ֱ���ֵķ�������������ֲ�����ͬʱ����˾�̬�����ģ�ͽ�������ؾ�������⡣
c. ʵ���˾�ȷ���Motif Finding����ķֲ�ʽ����ϵͳ��ʵ���������������ʵ�ֵķֲ�ʽ�㷨�����ϵͳ����Ч�������ʵ�������ڶ����ͬ����ʱ���нϺõ�Эͬ�ԡ�
�ο����ף�
[1] Krawetz S A, David D. Introduction to bioinformatics: A theoretical and practical approach[R]. Totowa: Humana Press, 2003.
[2] �� ϼ, �����, ����Ǫ, ��. ���������Žǻ�����ϵ�ǵ���9����ͻ���ȵ����ļ��[J]. ���ϴ�ѧѧ��: ҽѧ��, 2005, 30(5): 521-524.
SUN Xia, YIN Xin-zhen, WU Ling-qian, et al. Hotspot of the mutations of keratin 9 gene in a diffuse palmoplantar keratoderma family[J]. Journal of Central South University: Medicine Science, 2005, 30(5): 521-524.
[3] YU Jiang-sheng. NP completeness [R]. Beijing: Peking University, Institute of Computational Linguistics, 2003.
[4] Chin F, Leung H, Yiu S M, et al. Finding motifs for insufficient number of sequences with strong binding to transcription factor[C]//RECOMB04. San Diego: ACM Press, 2004: 125-132.
[5] Rajasekaran S, Balla S, Huang C H. Exact algorithms for planted motif problems[J]. Journal of Computational Biology, 2005, 12(8): 1117-1128.
[6] Shapiro J, Brutlag D. FoldMiner: Structural motif discovery using an improved superposition algorithm[J]. Protein Science, 2004, 13: 278-294.
[7] Hertz G Z, Stormo G D. Identifying DNA and protein patterns with statistically significant alignments of multiple sequences[J]. Bioinformatics, 1999, 15(7): 563-577.
[8] Lawrene C E, Altschul S F, Boguski M S, et al. Detecting subtle sequence signals: A Gibbs sampling strategy for multiple alignment[J]. Science, 1993, 262(5131): 208-214.
[9] Bailey T L, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers[C]//Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology. California: AAAI Press, 1994: 28-36.
[10] ������, Ī���. ����Internet��ͨ��ϵͳ����ʵ�黷�������ʵ��[J]. ���ϴ�ѧѧ��: ��Ȼ��ѧ��, 2006, 37(2): 128-133.
WANG Jian-xin, MO Qiu-ju. Design and implementation of communication system virtual environment based on Internet[J]. Journal of Central South University: Science and Technology, 2006, 37(2): 128-133.
[11] Pevzner P A, Sze S H. Combinatorial approaches to finding subtle signals in DNA sequences[C]//Proceedings of the 8th International Conference on Intelligent Systems for Molecular Biology. La Jolla: AAAI Press, 2000: 269-278.
[12] Sze S H, CHEN Jian-er. Find specific motifs in DNA sequences via cliques in k-partite graphs[R]. Texas A&M University: Departments of Computer Science and Biochemistry & Biophysics, 2003.
[13] Buhler J, Tompa M. Finding motifs using random projections[C]//Proceedings of the 5th Annual International Conference on Computational Molecular Biology. Montreal: ACM Press, 2001: 69-76.
[14] ���, ����־, ��ҫ��, ��. DNA������ģʽ���ֵ�һ�ֿ����㷨[J]. ��������ѧ��, 2005, 21(2): 121-129.
LI Dong-dong, WANG Zheng-zhi, DU Yao-hua, et al. A fast Motif Finding algorithm for dna sequence[J]. Acta Biophysica Sinica, 2005, 21(2): 121-129.
[15] Eskin E. Sparse sequence modeling with applications to computational biology and intrusion detection[D]. New York: Columbia University, 2002.
�ո����ڣ�2006-12-15�������ڣ�2007-01-08
������Ŀ��������Ȼ��ѧ����������Ŀ(60433020)
����飺����ƽ(1966-)���У����������ˣ���ʿ�����ڣ����²���������Ӧ�ü��������ݿ⼼���о�
ͨ�����ߣ�����ƽ���У����ڣ��绰��0731-8877936��E-mail: zpzhang@mail.csu.edu.cn
