

Density functional theory study of influence of impurity on electronic properties and reactivity of pyrite
LI Yu-qiong1, CHEN Jian-hua2, 3, CHEN Ye2, GUO Jin3
1. School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China;
2. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;
3. College of Physics Science and Engineering, Guangxi University, Nanning 530004, China
Received 2 April 2010; accepted 26 December 2010
Abstract: The electronic property of pyrite supercell containing As, Se, Te, Co or Ni hetero atoms were calculated using density functional theory (DFT), and the reactivities of pyrite with oxygen and xanthate were discussed by frontier orbital methods. The cell volume expands due to the presence of impurity. Co and Ni mainly affect the bands near Fermi levels, while As mainly affects the shallow and deep valence bands, and Se and Te mainly affect the deep valence bands. Electronic density analysis suggests that there exists a strong covalent interaction between hetero atom and its surrounding atoms. By frontier orbital calculation, it is suggested that As, Co and Ni have greater influence on the HOMO and LUMO of pyrite than Se and Te. In addition, pyrite containing As, Co or Ni is easier to oxidize by oxygen than pyrite containing Se or Te, and pyrite containing Co or Ni has greater interaction with collector. These are in agreement with the observed pyrite practice.
Key words: pyrite; impurity; density functional theory; electronic properties; reactivity
1 Introduction
Pyrite originating from different deposits and even from different sections of one deposit has different floatability, which is mainly ascribed to the variable properties of pyrite that has been observed. The heterogeneity mainly resulting from the presence of impurity elements in pyrite crystal leads to the variability in pyrite properties. The perfect pyrite, represented by the formula FeS2, is not encountered in natural specimens. ABRAITIS et al [1] reviewed the types of impurities found in natural pyrite. Their analysis showed that natural pyrite typically contained a variety of minor and trace elements which could exist as substitutions in the pyrite lattice or as inclusions.
Pyrite is a semiconducting mineral with narrow band gap (0.95 eV [2]) and its flotation process involves electrochemical reactions. It has been proved that, in the process of pyrite flotation, oxygen accepts electrons on the pyrite surface on which the cathodic reaction occurs, while xanthate loses electrons on the pyrite surface on which the anodic reaction occurs. The above process can be summarized by the following redox reactions:
Cathodic reaction O2+4e��2O2- (1)
Anodic reaction 2EX-��(EX)2+2e (2)
It has been confirmed that the rest potential of pyrite electrode and the equilibrium potential for the reaction (2) can affect the product forming on pyrite surface [3]. The presence of impurities can change the semiconductivity of pyrite and consequently influence the chemical reaction of pyrite electrode. LEHNER et al [4] and SAVAGE et al [5] studied the influence of arsenic, cobalt and nickel impurities on the electrical conductivity and semiconductivity type of pyrite. The crystal structure and electronic structure of pyrite can also be affected by the presence of impurities in pyrite. Using X-ray diffraction, FERRER et al [6] found that the pyrite lattice constant increases as Ni content increases. CHANDLER and BEN? [7] proposed the partial energy band scheme for cobalt and nickel impurities in pyrite by electron paramagnetic resonance (EPR). LEIGHTON et al [8] discussed the electronic structure of Co1-xFexS2 alloy. Using density functional method (DFT), BLANCHARD et al [9] suggested that there is a strong covalent bond between arsenic and iron atoms in pyrite, and the electronic structure of pyrite becomes metallic gradually with the increase of arsenic concentration.
Recently, CHEN et al [10-16] studied the effect of lattice impurities on electronic structures and flotation behaviors of sphalerite and galena. However, few theoretical results concerning the influence of impurities on the pyrite flotation behavior were published. In this study, the pyrite bearing Co, Ni, As, Se and Te impurities which are often incorporated into the pyrite lattice via a substitution mechanism is studied. To understand the effects of these impurities on the electronic structure and reactivity of pyrite would be helpful to solve the problems which might be encountered in pyrite flotation practice.
2 Computational details
Pyrite (FeS2) possesses a rocksalt type structure and belongs to the space group ![]() , with the Fe2+ cations on the corners and the face-centers positions of the cubic cell and the
, with the Fe2+ cations on the corners and the face-centers positions of the cubic cell and the ![]() dimmers occupying the anion sites along the ��111? directions (see Fig. 1(a)). The unit cell of pyrite contains four FeS2. Each Fe atom is coordinated to six S atoms, creating a distorted octahedron, while each S atom is coordinated to three Fe atoms and one S atom in a tetrahedral configuration. The 2��2��1 pyrite supercell with a Co or Ni atom substitution for a Fe atom or a As, Se or Te atom substitution for a S atom was used (see Fig. 1(b)).
dimmers occupying the anion sites along the ��111? directions (see Fig. 1(a)). The unit cell of pyrite contains four FeS2. Each Fe atom is coordinated to six S atoms, creating a distorted octahedron, while each S atom is coordinated to three Fe atoms and one S atom in a tetrahedral configuration. The 2��2��1 pyrite supercell with a Co or Ni atom substitution for a Fe atom or a As, Se or Te atom substitution for a S atom was used (see Fig. 1(b)).
Based on the density functional theory, the calculations were performed using the program CASTEP [17-18] and DMol3 [19-20]. The calculations of geometry optimization and electronic properties on pyrite were performed using CASTEP, GGA-PW91 [21], with plane wave cut-off energy of 270 eV and Monkhorst-Pack [22-23] k-point sampling density of 2��2��4. Only valence electrons were considered explicitly using ultrasoft pseudopotentials [24], and pseudo atomic calculations were performed for S 3s23p4��Fe 3d64s2��Co 3d74s2��Ni 3d84s2, As 4s24p3��Se 4s24p4��Te 5s25p4. The convergence tolerances for geometry optimization calculations were set to the maximum displacement of 0.002 ? , the maximum force of 0.08 eV/?, the maximum energy change of 2.0��10-5 eV/atom and the maximum stress of 0.1 GP, and the SCF convergence tolerance was set to 2.0��10-6 eV/atom. In addition, the spin-polarization was used for all calculations.
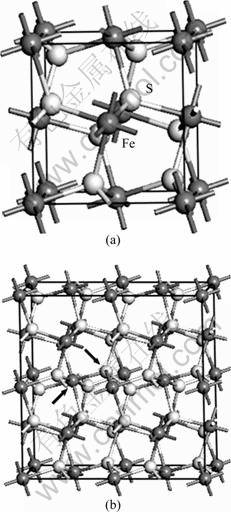
Fig. 1 Computed cell models of pyrite: (a) Pyrite unit cell; (b) 2��2��1 impurity-substituted pyrite supercell (Arrows indicate the impurity-substituted Fe and S positions)
The frontier orbitals of pyrite were calculated by DMol3 with a single-point energy method after optimization using CASTEP, while both the structure optimization and frontier orbital calculations on oxygen and dixanthogen were performed using DMol3. All the calculations were performed using DMol3, with GGA-PW91 method, DNP basis set, effective core potentials, a fine quality, and SCF convergence threshold of 1.0��10-6 eV/atom.
3 Results
The substitution energy refers to the energy required when impurity atom substitutes for matrix atom. Here the substitution energy of an impurity in the pyrite lattice,![]() , is defined as follows according to NISHIDATE et al [25]:
, is defined as follows according to NISHIDATE et al [25]:
![]() (3)
(3)
where ![]() and
and ![]() are the total energies of impurity-substituted and perfect pyrites, respectively. Ex and Eimpurity are defined as the calculated total energies of the substituted matrix atom (x= Fe or S) and the impurity atom (impurity = Co, Ni, As, Se or Te), respectively. The smaller the value of ?E is, the more easily the substitution reaction will occur, i.e. the impurity will exist in pyrite lattice more easily.
are the total energies of impurity-substituted and perfect pyrites, respectively. Ex and Eimpurity are defined as the calculated total energies of the substituted matrix atom (x= Fe or S) and the impurity atom (impurity = Co, Ni, As, Se or Te), respectively. The smaller the value of ?E is, the more easily the substitution reaction will occur, i.e. the impurity will exist in pyrite lattice more easily.
The impurity substitution energies were calculated by allowing the pyrite cell volume to relax. The calculated results are listed in Table 1, which demonstrates that the substitution energies of five impurities are all positive, indicating that these impurities cannot spontaneously enter into pyrite lattice at T=0 K. However, in practice these impurities may easily enter into pyrite lattice in the process of crystallization under high temperature and pressure.
Table 1 Substitution energy of impurity in pyrite lattice, and cell volume of perfect and impurity-substituted pyrites
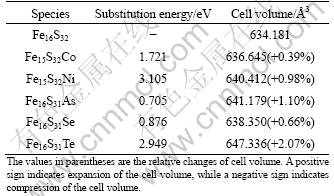
The calculated lattice parameter a0=5.4121 ? deviates from the experimental value a0=5.4166 ? [26] only by about 0.1%, suggesting the reliability of the calculation. The incorporation of five impurities results in a variable expansion of the lattice (see Table 1). Our calculated results are consistent with other works[6, 9, 27]. The extent of lattice expansion could be explained by the different covalent radii of atoms and the different bond lengths between bonding atoms. The extent of lattice expansion resulting from the impurity increases in the following order: Co< Se< Ni<>
Figures 2 and 3 show the band structure and partial DOS of pyrite when the zero of energy was set at the Fermi level (EF). Our calculations are in good agreement with other works [28-34]. The band structure is split into five groups of bands between -17 and 5 eV. The two groups between -17 and -10 eV have almost entirely the character of S 3s states, only with few contributions from Fe 3d and S 3p states. The band in the range from -7.5 to -1.5 eV below the valence band maximum (VBM) is formed of hybridized S 3p and Fe 3d eg states with the main contribution from S 3p. The band just below Fermi level is formed of S 3s and non-bonding Fe 3d t2g states with the main contribution from Fe 3d state. Finally, the conduction band is mainly formed of hybridized anti-bonding S 3p and Fe 3d ![]() states with few contributions from S 3s states. In addition, the contribution from Fe 4s is very small and not shown in the figure.
states with few contributions from S 3s states. In addition, the contribution from Fe 4s is very small and not shown in the figure.
Co and Ni impurities substitution for Fe introduces bulk defect states within the band gap (see Figs. 2(b) and 2(c) and Figs. 3(b) and 3(c)). This is consistent with the study by CHANDLER and BEN? [7]. Co and Ni would bond with S atom in pyrite crystal, forming covalent Co��S and Ni��S bonds. Electronic structure calculations by BULLETT [28] showed that the total DOS of CoS2 and NiS2 shifts to lower energy in contrast with that of FeS2. Our calculations showed that the DOS of pyrite shifts to lower energy by about 1 eV due to the incorporation of Co or Ni, suggesting that the Co- and Ni-bearing pyrites are more oxidative. This is helpful to the xanthate oxidation and then adsorption at pyrite surface. It was shown that there exist new state peaks in the band gap (at 0.2 eV and 0) generated from Co 3d and Ni 3d states, respectively, and also new state peaks at -2.3 eV generated from their 3d states, respectively.
As impurity introduces new state peaks at -10 and -13.7 eV generated from its 4s state, respectively. In addition, there exists a new state peak at -1.5 eV generated from As 4p state (Fig. 2(d) and Fig. 3(d)). The DOS of Se-substituted pyrite is most similar to that of perfect pyrite (Fig. 2(e) and Fig. 3(e)), and there is no apparent peak of impurity state in the band. This is relative to more similar properties of Se to S reflected in electronegativity and covalent radii than that of the others to S. Te impurity introduces new state peaks at -10.8 and -14 eV generated from its 5s state (see Fig. 2(f) and Fig. 3(f)).
The bonding between atoms can be clearly shown by the electron density map. The presence of strong As-Fe, Se-Fe, Te-Fe, Co-S, and Ni-S hybridization interactions is clearly visible on total electron density maps (see Fig. 4).
Calculations on perfect pyrite produce a low-spin state, in line with experimental findings. At the 2.08% impurity concentration, the calculations on Se-, Te-, Co-, or Ni-substituted pyrite produce low-spin states, while on As-substituted pyrite produces a spin-polarized state (see Fig. 5). Spin DOS near Fermi level is mainly generated from Fe 3d state. By contrast with spin-neutral pyrite, spin-polarized pyrite would be more reactive to magnetic specie such as O2 than non-magnetic species��hence As-substituted pyrite would be oxidized easily, which is consistent with the fact that pyrite containing As is oxidized easily in nature.
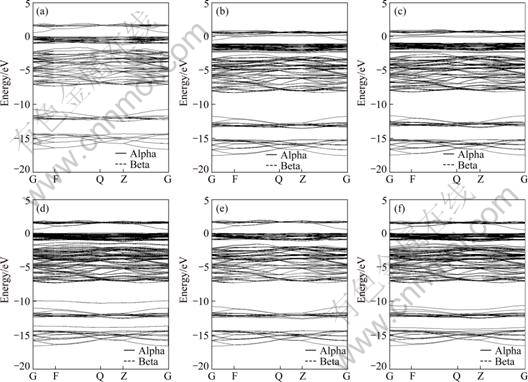
Fig. 2 Band structures of perfect pyrite and impurity-substituted pyrite: (a) Perfect pyrite; (b) Co-substituted pyrite; (c) Ni-substituted pyrite; (d) As-substituted pyrite; (e) Se-substituted pyrite; (f) Te-substituted pyrite (The zero of energy was set at the Fermi level. Alpha and beta indicates the spin up and spin down, respectively)
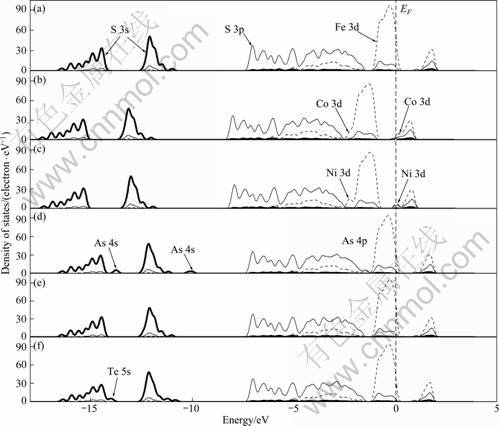
Fig. 3 DOS of perfect pyrite and impurity-substituted pyrite: (a) Perfect pyrite; (b) Co-substituted pyrite; (c) Ni-substituted pyrite; (d) As-substituted pyrite; (e) Se-substituted pyrite; (f) Te-substituted pyrite (The zero of energy was set at the Fermi level)
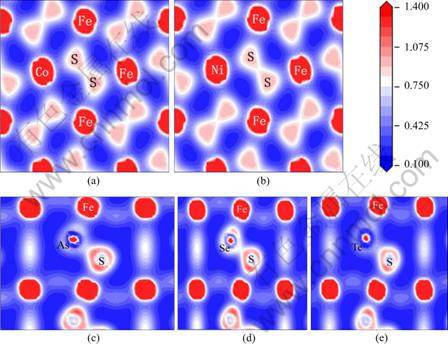
Fig. 4 Electron density maps of pyrite supercell containing impurity: (a) Co-substituted pyrite; (b) Ni-substituted pyrite; (c) As-substituted pyrite; (d) Se-substituted pyrite; (e) Te-substituted pyrite
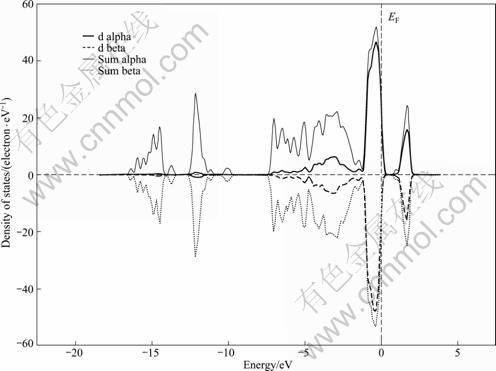
Fig. 5 Spin density of states of As-substituted pyrite
The presence of impurities may vary the semiconductivity type of sulfide minerals, resulting in both p-type and n-type, and even p-n-type semiconductivity [35]. The calculated results suggested that Co- and Ni-substituted pyrites exhibit n-type semiconductivity, while As-, Se-, and Te-substituted pyrites exhibit p-type semiconductivity. S, Se and Te in the same group in period table, who adopt the outer electron configurations of s2p4, were studied similarly in behaviors in metal-matte systems by SCHLITT and RICHARDS [36]. PRIDMORE and SHUEY [37] indicated that As is an electron acceptor impurity in pyrite, and high As content in pyrite has been found to impart p-type properties. LEHNER et al [4] found that the carries tend to be holes in the presence of As in pyrite, and this pyrite is p-type, while the carries tend to be electrons in the presence of Co or Ni, and these pyrites are n-type. FERRER et al [6] also found that Ni-doped pyrite is an n-type semiconductor. In addition, our calculations showed that the energy gaps of As-, Co-, and Ni-substituted pyrites are indirect, while the energy gaps of Se- and Te-substituted pyrite are direct.
The above results suggested that the presence of impurities could slightly change the crystal structure, electronic structure, semiconductivity type, and internal covalent bonding in pyrite. Co and Ni mainly affect the bands near Fermi levels, while As mainly affects the shallow and deep valence bands, and Se and Te mainly affect the deep valence bands with introducing new impurity energy levels. Co and Ni enter the pyrite lattice as donors, resulting in n-type semiconductivity, while As, Se and Te enter the pyrite lattice as acceptors, resulting in p-type semiconductivity. All the evidences showed that the properties of pyrite are changed in the presence of impurities, and consequently the reactivity of pyrite with flotation reagents will be affected.
The extent of the interaction of pyrite with reagents is inversely proportional to the energy difference between the HOMO (highest occupied molecular orbital) which donates electrons and LUMO (lowest unoccupied molecular orbital) which accepts electrons [38], and the magnitude of the atomic orbital coefficient, which indicates the contribution of atoms to frontier orbital, also plays an important part in this interaction.
A high value of coefficient (absolute value) indicates a large contribution of atom to the frontier orbitals, while a low value (absolute value) indicates a small contribution of atom. Moreover, the same signs of coefficient denote the bonding between atoms, while the opposite signs of coefficient denote the anti-bonding state between atoms. Here only the absolute value and maximum value are concerned. Table 2 shows the atomic orbital coefficients of HOMO and LUMO. It is shown that the coefficient of Fe atom (0.238) is much larger than that of S atom (0.068) for HOMO, indicating that the main contribution of HOMO for perfect pyrite is generated from Fe atom, which is in accordance with the fact that Fe atom in pyrite is easily oxidized from Fe2+ to Fe3+. While the coefficient of S atom (0.124) is much larger than that of Fe atom (0.004) for LUMO orbital, indicating that the main contribution of LUMO of perfect pyrite is generated from S atom.
Table 2 Atomic orbital coefficients of HOMO and LUMO of perfect and impurity-substituted pyrites

Co and Ni impurities have similar influence on the frontier orbital. The Co and Ni have great effect on LUMO, whose coefficients are up to 0.421 and 0.447 respectively, indicating that Co and Ni will directly take part in the oxidation reaction of LUMO. In addition, the presence of Co and Ni impurities results in the increase of coefficients of Fe atom (0.202 and 0.191, respectively) and S atom (0.329 and 0.342, respectively). The coefficient of Fe atom in HOMO is decreased in the presence of Co and Ni.
Arsenic impurity only influences the HOMO of pyrite, whose coefficient is up to 0.315, and the coefficients of Fe and S in HOMO are also enhanced. These results suggest that the pyrite can be oxidized easily in the presence of As impurity, and As impurity will participate the oxidation reaction. This is the reason why As is easily dissolved out from natural pyrite surface. Se impurity has little influence on the HOMO and LUMO of pyrite, which is mainly ascribed to the similar chemical properties between Se and S atoms. Te impurity slightly increases the coefficients of Fe and S atoms in HOMO and that of S atom in LUMO.
Based on the frontier orbital theory, in the pyrite oxidation, the reaction should occur between the LUMO of oxygen and the HOMO of pyrite. The extent of interaction between HOMO and LUMO can be defined by the absolute energy difference (|��E1|) between them. The |��E1| is defined as:
|��E1|=|![]() | (4)
| (4)
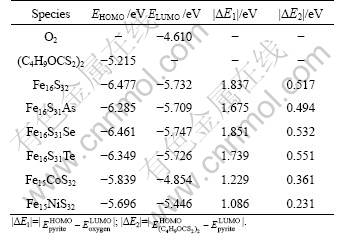
The small |��E1| indicates strong interaction. The calculated values of the HOMO and LUMO energies and the absolute difference (|��E1|) are shown in Table 3. It is shown that As, Co and Ni lower the |��E1|, indicating that the presence of As, Co and Ni can enhance the oxidation of pyrite, which is consistent with the observed pyrite oxidation [39]. Se and Te impurities do not apparently change the |��E1|, suggesting that Se and Te would not affect the oxidation of pyrite.
During froth flotation, xanthate is oxidized to dixanthogen on pyrite surface and forms hydrophobic surface, and then pyrite is collected by attaching to rising air bubbles. The frontier orbital of butyl-dixanthogen ((C4H9OCS2)2) was calculated. Based on the frontier orbital theory, the reaction should occur between the LUMO of pyrite and the HOMO of (C4H9OCS2)2. The extent of the interaction between HOMO and LUMO can be defined by the absolute energy difference (|��E2|) between them. The |��E2| is defined as:
|��E2|=|![]() | (5)
| (5)
The calculated values of the HOMO and LUMO energies and the absolute difference (|��E2|) are shown in Table 3 also. It is shown that for perfect pyrite the |��E2| is small (0.517 eV), suggesting strong interaction between perfect pyrite and butyl-dixanthogen. The incorporation of Co and Ni impurities significantly lowers the |��E2| (0.361 eV for Co-bearing pyrite and 0.231 eV for Ni-bearing pyrite), while As, Se and Te impurities do not influence the |��E2| apparently. It can be speculated that Co and Ni would greatly enhance the interaction of pyrite with butyl-dixanthogen, while As, Se and Te would have little effects on the interaction of pyrite with butyl-dixanthogen. This is in agreement with the processing practice of pyrite that the pyrite floatability typically gets better due to the presence of Co or Ni.
Table 3 HOMO and LUMO orbital energy of perfect, impurity-substituted pyrites, and O2 and (C4H9OCS2)2, and absolute value of orbital energy difference between pyrite and either O2 or (C4H9OCS2)2.
4 Conclusions
1) The presence of Co, Ni As, Se and Te impurities makes the expansion of pyrite lattice, and the extent of the expansion increases in the order: Co<><><>
2) Co and Ni mainly affect the electronic structures near Fermi levels, while As mainly affects the electronic structure at shallow and deep valence bands, and Se and Te impurities mainly affect the electronic structures at deep valence bands of pyrite.
3) At the impurity concentration of 2.08%(mole fraction), Co-, Ni-, Se- and Te-substituted pyrites are predicted to be spin-neutral, while As-substituted pyrite is predicted to be spin-polarized.
4) The interaction of pyrite containing different impurity with oxygen and xanthate were studied using frontier orbital method. The presence of As, Co and Ni can enhance the oxidation of pyrite, while Se and Te would not affect the oxidation of pyrite. Co and Ni can greatly enhance the interaction of pyrite with butyl-dixanthogen, while As, Se and Te have little effects on the interaction of pyrite with butyl-dixanthogen.
References
[1] ABRAITIS P K, PATTRICK R A D, VAUGHAN D J. Variations in the compositional, textural and electrical properties of natural pyrite: A review [J]. International Journal of Mineral Processing, 2004, 74(1-4): 41-59.
[2] SCHLEGEL P, WACHTER P. Optical properties, phonons and electronic structure of iron pyrite (FeS2) [J]. Journal of Physics C: Solid State Physics, 1976, 9(17): 3363-3369.
[3] ALLISON S A, GOOLD L A, NICOL M J, GRANVILLE A. A determination of the products of reaction between various sulfide minerals and aqueous xanthate solution, and a correlation of the products with electrode rest potentials [J]. Metallurgical and Materials Transactions B, 1972, 3(10): 2613-2618.
[4] LEHNER S W, SAVAGE K S, AYERS J C. Vapor growth and characterization of pyrite (FeS2) doped with Co, Ni, and As: Variations in semiconducting properties [J]. Journal of Crystal Growth, 2006, 286(2): 306-317.
[5] SAVAGE K S, STEFAN D, LEHNER S W. Impurities and heterogeneity in pyrite: Influence on electrical properties and oxidation products [J]. Applied Geochemistry, 2008, 23(2): 103-120.
[6] FERRER I J, HERAS C D L, S?NCHEZ C. The effect of Ni impurities on some structural properties of pyrite thin films [J]. Journal of Physics: Condensed Matter, 1995, 7(10): 2115-2121.
[7] CHANDLER R N, BEN? R W. EPR study of the solid solutions NixFe1-xS2, CoxFe1-xS2, and CoxNiyFe1-x-yS2 [J]. Physical Review B, 1973, 8(11): 4979-4988.
[8] LEIGHTON C, MANNO M, CADY A, FREELAND J W, WANG L, UMENOTO K, WENTZCOVITCH R M, CHEN T Y, CHIEN C L, KUHNS P L, HOCH M J R, REYES A P, MOULTON W G, DAHLBERG E D, CHECKELSKY, J, ECKERT J. Composition controlled spin polarization in Co1-xFexS2 alloys [J]. Journal of Physics: Condensed Matter, 2007, 19(31): 315219-1-315219-22.
[9] BLANCHARD M, ALFREDSSON M, BRODHOLT J, WRIGHT K, CATLOW C R A. Arsenic incorporation into FeS2 and its influence on dissolution: A DFT study [J]. Geochimica et Cosmochimica Acta, 2007, 71(3): 624-630.
[10] CHEN J H, CHEN Y. A first-principle study of the effect of vacancy defects and impurities on the adsorption of O2 on sphalerite surfaces [J]. Colloids and Surfaces A, 2010, 363(1-3): 56-63.
[11] CHEN Y, CHEN J H. The first-principle study of the effect of lattice impurity on adsorption of CN on sphalerite surface [J]. Minerals Engineering, 2010, 23(9): 676-684.
[12] CHEN J H, CHEN Y, LI Y Q. Quantum-mechanical study of effect of lattice defects on surface properties and copper activation of sphalerite surface [J]. Transactions of Nonferrous Metals Society of China, 2010, 20(6): 1121-1130.
[13] CHEN Y, CHEN J H, GUO J. A DFT study on the effect of lattice impurities on the electronic structures and floatability of sphalerite [J]. Minerals Engineering, 2010, 23(14): 1120-1130.
[14] CHEN J H, WANG L, CHEN Y, GUO J. A DFT study of the effect of natural impurities on the electronic structure of galena [J]. International Journal of Mineral Processing, 2011, 98(3-4): 132-136.
[15] CHEN J H, ZENG X Q, CHEN Y, ZHANG H P. First-principle theory calculations of electronic structure of sphalerite with vacancy and impurity [J]. The Chinese Journal of Nonferrous Metals, 2010, 20(4): 765-771. (in Chinese).
[16] CHEN J H, CHEN Y, ZENG X Q, LI Y Q. First principle study of effect of Fe impurity on electronic structure and activation of sphalerite surface [J]. The Chinese Journal of Nonferrous Metals, 2009, 19(8): 1517-1523. (in Chinese).
[17] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods using CASTEP [J]. Zeitischrift fuer Kristallographie, 2005, 220(5-6): 567-570.
[18] SEGLL M D, LINDAN P J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: ideas, illustrations and the CASTEP code [J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[19] DELLEY B. An all-electron numerical method for solving the local density functional for polyatomic molecules [J]. Journal of Chemical Physics, 1990, 92(1): 508-517.
[20] DELLEY B. From molecules to solids with the DMol3 approach [J]. Journal of Chemical Physics, 2000, 113(18): 7756-7764.
[21] PERDEW J P, CHEVARY J A, VOSKO S H, JACHSON K A, PEDERSON M R, SINGH D J, FIOLHAIS C. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation [J]. Physical Review B, 1992, 46(11): 6671-6687.
[22] MONKHORST H J, PACK J D. Special points for brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188-5192.
[23] PACK J D, MONKHORST H J. Special points for Brillouin-zone integrations��A reply [J]. Physical Review B, 1977, 16(4): 1748-1749.
[24] VANDERBILT D. Soft self-consistent pseudopotentials in generalized eigenvalue formalism [J]. Physical Review B, 1990, 41(11): 7892-7895.
[25] NISHIDATE K, YOSHIZAWA M, HASEGAWA M. Energetics of Mg and B adsorption on polar zinc oxide surfaces from first principles [J]. Physics Review B, 2008, 77(3): 035330-1-035330-6.
[26] PRINCE K C, MATTEUCCI M, KUEPPER K, CHIUZBAIAN S G, BARKOWSKI S, NEUMANN M. Core-level spectroscopic study of FeO and FeS2 [J]. Physical Review B, 2005, 71(8): 085102-1��085102-9.
[27] SAVAGE K S, TINGLE T N, O'DAY P A, WAYCHUNAS G A, BIRD D K. Arsenic speciation in pyrite and secondary weathering phases, Mother Lode Gold District, Tuolumne County, California [J]. Applied Geochemistry, 2000, 15(8): 1219-1244.
[28] BULLETT D W. Electronic structure of 3d pyrite- and marcasite-type sulphides [J]. Journal of Physics C: Solid State Physics, 1982, 15(30): 6163-6174.
[29] EDELBRO R, SANDSTR?M ?, PAUL J. Full potential calculations on the electron bandstructures of sphalerite, pyrite and chalcopyrite [J]. Applied Surface Science, 2003, 206(1-4): 300-313.
[30] OERTZEN G U, SKINNER W M, NESBITT H W. Ab initio and X-ray photoemission spectroscopy study of the bulk and surface electronic structure of pyrite (100) with implications for reactivity [J]. Physical Review B, 2005, 72(23): 235427-1��235427-10.
[31] OERTZEN G U, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides [J]. Physics and Chemistry of Minerals, 2005, 32(4): 255-268.
[32] WOMES M, KARNATAK R C, ESTEVA J M, LEFEBVRE I, ALLA G, OLIVIER-FOURCADE J, JUMAS J C. Electronic structures of FeS and FeS2: X-ray absorption spectroscopy and band structure calculations [J]. Journal of Physics and Chemistry of Solids, 1997, 58(2): 345-352.
[33] OPAHLE I, KOEPERNIK K, ESCHRIG H. Full potential band structure calculation of iron pyrite [J]. Computational Materials Science, 2000, 17(2-4): 206-210.
[34] ZHAO G L, CALLAWAY J, HAYASHIBARA M. Electronic structures of iron and cobalt pyrites [J]. Physical Review B, 1993, 48(21): 15781-15786.
[35] FAVOROV V A, KRASNILOV V J, SYCHUGOV V S. Variations in semiconductor properties of pyrite and arsenopyrite and their determinants [J]. International Geology Review, 1974, 16(4): 385-394.
[36] SCHLITT W J, RICHARDS K J. The behavior of selenium and tellurium in metal-matte systems [J]. Metallurgical and Materials Transactions, 1973, 4(3): 819-825.
[37] PRIDMORE D F, SHUEY R T. The electrical resistivity of galena, pyrite, and chalcopyrite [J]. American Mineralogist, 1976, 61(3-4): 248-259.
[38] SAUER J, SUSTMANN R. Mechanistic aspects of diels-alder reactions: A critical survey [J]. Angewandte Chemie International Edition in English, 1980, 19(10): 779-807.
[39] LEHNER S, SAVAGE K. The effect of As, Co, and Ni impurities on pyrite oxidation kinetics: Batch and flow-through reactor experiments with synthetic pyrite [J]. Geochimica et Cosmochimica Acta, 2008, 72(7): 1788-1800.
���ʶԻ�����������ʼ�
��Ӧ����Ӱ����ܶȷ��������о�
������1, �½���2, 3, �� ��2, �� ��3
1. ������ѧ ��ѧ����ѧԺ������ 530004��
2. ������ѧ ��Դ��ұ��ѧԺ������ 530004��
3. ������ѧ ������ѧ�빤�̼���ѧԺ������ 530004
ժ Ҫ��
�����ܶȷ������ۼ��㺬���顢�����ڡ��ܻ��������ʵĻ�����Ľṹ�͵������ʣ�������ǰ�߹���������ۺ����ʻ������������ͻ�ҩ�ķ�Ӧ���ԡ����ʵĴ���ʹ������������͡��ܺ�����Ҫ�Է����ܼ��������ܴ�����Ӱ�죬����������Ҫ�Ի�����dz������۴�����Ӱ�죬��������ҪӰ����۴�������ܶȷ���������������е�����ԭ�Ӷ�������Χ��ԭ���γɽ�ǿ�Ĺ�������á�ǰ�߹������������顢�ܺ������ʶԻ������HOMO��LUMO��Ӱ������������ʴ��⣬���顢�ܻ����Ļ�����Ⱥ������ڵĻ���������ױ����������������ܻ����Ļ��������ҩ���ռ������ø�ǿ����������۲쵽�Ļ�����ʵ����������
�ؼ��ʣ�
���������������ܶȷ���������������������Ӧ������
(Edited by YUAN Sai-qian)
Foundation item: Project (50864001) supported by the National Natural Science Foundation of China
Corresponding author: CHEN Jian-hua; Tel: +86-771-3232200; E-mail: jhchen1971@sina.com
DOI: 10.1016/S1003-6326(11)60946-1
