
���±��: 1004-0609(2005)07-1069-06
Al-Li�Ͻ�ʱЧ���ڵļۼ�����
��Ӣ��1, 2, �ƴ���1, Ī���1, ��־ǿ1, �� ��1, Τ����1
(1. ������ѧ ������ѧ�빤�̼���ѧԺ, ���� 530004; 2. �й���ѧԺ ���ʲ�����������, ���� 110016)
ժҪ: ���ù��徭���������(EET), ��Al-Li�Ͻ�ʱЧ���ڵ�����ƫ�۾����ļ۵��ӽṹ�����˼��㡣 ����������: ��������λ��ƫ�۾����ļ�����ǿ��ΪAl��Al��, ����Alԭ�ӵĹ��۰뾶��Liԭ�ӵĹ��۰뾶Ҫ��; ������λ��ƫ�۾�������ǿ��ΪAl��Li��, Alԭ�ӵĹ��۰뾶Ҫ��Liԭ�ӵĹ��۰뾶ҪС; �ڿ�λ���ڵ������, ����Alԭ����Liԭ�ӵĵ縺���������, ��ʹAl��Liԭ�ӽ��, �����γ�Al-Li�̳���ṹƫ����, ���ֺ���λ�Ķ̳���ṹ�ܿ��ܾ��Ǧġ�(Al3Li)�������ǰ�ṹ��������̥; ����Al-Li-��λ����ƫ�۾�����Al��Li����Ȼ������Ҫǿ����, ���, �������кϽ����ɵ�Al-Li-��λƫ�۾����ԺϽ��������������Ҫǿ������; ���������Ħġ�(Al3Li)��������������������, ����ǿ���������; ����Al3Li����干��, �����������ɢ���������������������ǿ��, ͬ���ԺϽ����ǿ�����á�
�ؼ���: Al-Li�Ͻ�; Al3Li; ��λ; �۵��ӽṹ; ��ѧ���� ��ͼ�����: TG111.1
���ױ�ʶ��: A
Calculation on valence electronic structures of Al-Li alloy under earlier aging condition
GAO Ying-jun1, 2, HUANG Chuang-gao1, MO Qi-feng1, LAN Zhi-qiang1, LIU Hui1, WEI Yin-yan1
(1. School of Physics Science and Technology, Guangxi University, Nanning 530004, China;
2. Center of International Materials and Physics, Chinese Academy of Sciences, Shenyang 110016, China)
Abstract: The valence electron structures of the segregated cell of Al-Li alloy in earlier aging condition were calculated according to the empirical electron theory (EET) in solid. The results show: the strongest bond is the Al-Al bond in the segregated cell without containing vacancy, where the Al atomic covalence radius is greater than that of Li atom in the cell; while the strongest bond is the Al-Li bond in the segregated cell containing vacancy, and the Al atomic covalence radius in the cell is less than that of Li atom. Since the difference of electronagativity between the Al and Li atoms is obvious, it is inclined to formed the Al-Li segregated cell of short range order structure in the condition of vacancy present. The short range order structure containing vacancy is probably the embryo or precursor structure of the metastable phase �ġ�(Al3Li). Because the strongest covalent bond in the Al-Li-vacancy segregated cell in alloy formed in quenching is the main strength reason for supersaturated solid solution of alloy. The bond net of succeeding precipitation of �ġ�(Al3Li) has the picture of anisotropic Al-Al bonding and the bond intensity enhanced. Since the �ġ�(Al3Li) is coherence with matrix, the bond net strength is enhanced by the precipitation of �ġ�(Al3Li) and so strengthen the alloy.
Key words: Al-Li alloy; Al3Li; vacancy; covalence bond; mechanical properties
Al-Li�Ͻ���е��ܶ�, �߱�ǿ�Ⱥͱ�ģ��, ���õ���ʴ�Ժ�����ĵ������Ե��ص�[1-3], �ѳ�Ϊ���պ������Ҫ�ṹ���ϡ� ���ھ����ܴ��ʱЧ��Al-Li�Ͻ�, ͨ����Ϊ��������������������干��Ħġ�(Al3Li)������, ����ʱЧʱ����ӳ�, ��������ȶ��Ħ�(AlLi)ƽ���ࡣ ���������, ʵ���о�[4-7]����Li��������5.5%(Ħ������)��Al-Li�Ͻ���ܴ��, �õ��Ĺ���������, ������ʱЧ���������Ȧġ�(Al3Li)��֮ǰ, �ڼ���ʱ���ںϽ�Эͬ����Spinodal�ֽ��������[8, 9], �����γ�����G.P��������ԭ�Ӷ̳������ǰ������ԭ��ƫ�۽ṹ��, Ȼ�����ݻ��γ�����Ħġ�(Al3Li)�����ࡣ һЩ����[10-14]��������Ӧ�õ�����������͵ͳ�HallЧӦ�����������̬�Ͻ������ԭ�ӺͿ�λ�ԺϽ�ĵ����Hallϵ����Ӱ��, ��ʾ������ԭ�����λ��ʱЧ�Цġ����γɵ����á� ��Щǰ��ԭ��ƫ�۽ṹ���ݻ����ܹ�ʣ����λ�Ŀ���[6,15], ��λ��״̬����Щԭ��ƫ�����ṹ���γ�����������Ҫ�����á� ʵ���о�[2]������, Al-Li�Ͻ������еĸߵ���ģ������Liԭ�Ӽ�����Χԭ�ӳɼ��ļ۵��ӽṹ���Ž��ܵ���ϵ, ��Ҳ��һ��˵����ʱЧ����������Al��Liԭ�ӵĵ縺�Բ�����ɵĻ���������ṹ, �ı��˺Ͻ��ڲ���ԭ�ӳɼ�״̬, �Ӷ�����Ͻ����ܷ����ı䡣 ����, ���Ǹ�ע�شӼ۵��ӽṹ��ν�ʾ�Ͻ���е�����������ܵ�����ԭ��
��Щ������ⶼ��ѧ�ߴ����ۺ�ʵ���϶�Al-Li�Ͻ�ļ۵��ӽṹ�������о�[16, 17]�� �������õ�һԭ��������Al-Li�Ͻ�ļ۵��ӽṹ����, ȡ����������Ľ��, ��������̽�Ϊ���ӡ� ���ڼۼ�����[18]���ܴ����۽����Ĺ��徭���������(EET)[19], ���ṩ��һ������������ϵ�۵��ӽṹ�ļ�ݼ��㷽����������� (BLD)��, �ɹ������ںϽ��ԭ��ƫ����Ͻ�����о�[20], ʹ���о��Ͻ�ĺ�����ܿ����ݵ��Ͻ�ԭ�ӵļ۵��ӽṹ���, Ϊ�Ͻ��������ṩ�����ε�����ָ��[20, 21]�� ������������EET����, ����Al-Li���ܴ��̬�Ͻ�ʱЧ���ںϽ��ڲ�ԭ�ӵļ۵��ӳɼ�״̬, �Ӽ۵��ӽṹ��η���Al-Li�Ͻ�ʱЧ�����ܵ�Ӱ�졣
1 ����ģ��
����Al-Li�Ͻ���ͼ��Al�˵Ħ��ྭ���¹��ܱ�ˮ����, ���γɹ����͵�Li���ʹ����塣 ���ͬʱ, ����Liԭ�����λ��ǿ�Ľ������, �ڹ������н����γɴ����Ĺ����Ϳ�λȱ�ݡ� ��Al-Li�Ͻ���Li��������(4%��x��12%)�������, ���ǵ�Al��Liԭ��֮����д�Ļ��ϼ�֮�������Ļ�ѧ������, ͬʱLiԭ�ӶԿ�λ�н�ǿ�ķ�������[11], ���, ��Al-Li�Ͻ���ܴ��������н�����־ֲ��۲������ԡ� ��Al-Li�Ͻ��������۲������Կ����á����ƫ�۾�����ģ��[20]���������� ����ģ����Ϊ, �ںϽ����״̬, Al-Li������ɿ����ɼ���ƫ�۾�����϶�������: 1) ��Al����(FCC����); 2) ��Li��Al����(FCC����), ��Liԭ������˴�Al�����IJ�������Alԭ��; 3) ���ǵ��Ͻ���ܴ���������, ���д����ķ�ƽ���λ����, ��Liԭ�����λ���к�ǿ�Ľ������[11], ��Щ��ʣ��λ��Liԭ�ӷ���, ���γ�Liԭ��-��λ��, ����Al-Li�������л�Ӧ�ð�����Liԭ�����λ��������γɵ�Al-Li-��λ��ƫ�۾����� �⼸��ƫ�۾������Ͻ�Ĺ��ܴ��, ���ɱ����̬�Ͻ𱣴�����, ��Щ�ṹ���ԺϽ�����ܲ�����Ҫ��Ӱ�졣
Ϊ�о��ͼ��㷽��, ����������[2, 6]�е�ʵ�������бȽ�, ������Al-8%Li(Ħ������)�Ͻ���Ϊ�о����� ��������LiŨ�ȵĺϽ�, ���Դ��ԵĹ������Li��Al-Liƫ�۾��������ƽ�������ԼΪ2~3��ԭ�Ӽ�ࡣ ���۵��¶�(875K)������Al�Ͻ������, ����[22]����������Ŀ�λŨ�ȿɴ�10-3�� ��ʱ, ���Դ��ԵĹ��Ƴ�Al-Li�������п�λ��ƽ�������10��ԭ�ӵľ������ҡ� ���, ���Խ�����ΪAl-Li-��λ����֮���ƽ������Ϊ6~7��ԭ�Ӽ�ࡣ ��˿ɼ�, �����о���Al-8%Li�Ͻ����, ����λ��Al-Li-��λ��ƫ�۾���������Ҫ�Ȳ�����λ��Al-Li��ƫ�۾����������Ҫ�١� ��������[6]ָ����Al-Li���Ͻ���Эͬ����, ���, ��������Ǿ��жԳ��Ծ�������Ǻ����ġ� ���ڲ�����λ��Al-Li����, Ϊ�о��������ֻ�������ֲ�ͬ�ɷ����͵ĶԳ��Ծ���, ���纬Li�ֱ�Ϊ6.25%, 12.5%, 25%(Ħ������)��Al-Li������ ��3�ֺ���ͬLi�ɷֵľ����ֱ���Al-6.25%Li�� Al-12.5%Li�� Al-25%Li��ʾ, ��ͼ1(c), ͼ2(b), ͼ1(b)��ʾ�� ���ǵ�Liԭ�����λ�н�ǿ�Ľ����, ���γ�Liԭ��-��λ��, ���ں���λ��Al-Li-��λ����ƫ�۾�����, Liԭ�ӺͿ�λӦ����ռ�����ĵ�λ��, �Ա�ʹLiԭ�����λλ�þ����ؿ����� ѡȡ�⼸�ֶԳƾ����ṹģ����Ϊ�о�����, ��Ҫ����������[14]������Al-Li�Ͻ�߷ֱ�羵��Ƭ�� ����Ƭ��ʾ���Ͻ���̬��ǰ�ṹ���ж̳�����ṹ����ΪL12�ͽṹ, �����ȶ��Բ���Al3Li����ṹ��, ������Щǰ�ṹ�����ı߽�[8, 9]��ģ���� ��Щģ���ı߽���������ڴ��ڿ�λ�š� λ���� λ������ȱ�ݽṹ�� ����ǵ�Al-Li-��λ�����������������ṹ��ͼ2(a)��ʾ��
��������[22]ָ�����������������Li�����Ӷ��½�, ��˵���˺�Li��ƫ�۾����ľ�����С�ڲ���Li�Ĵ�Al�����ľ������� ʵ���õĵ�����Ϊ�����־����ߴ�ļ�Ȩƽ��ֵ�� ��֪��Al�ľ�����[22]Ϊa0 =0.40496nm, ������ʵ����ɿ����Ƴ���8%Li��Al-Li�Ͻ�ĵ�����Ϊa=0.4046nm, ��Al-Li��������, ��Ħ������Ϊ8%ʱ, ƽ��������Ϊa[TX-]= 0.4046nm, �����������Li��Al-Liƫ�۾����ľ�����Ϊa1=0.40434nm�� Ϊ���㷽�����, Al-Li-��λ��ƫ�۾����ľ��������Ƶ�ȡAl-Liƫ�۾����ľ���������, ������λ�Ծ�������Ӱ��ͨ��Al��Liԭ�ӵ��ӽױ仯����ӳ�� �ڹ�������ʱЧ������, ���ǵ�������������干������Ȧġ�(Al3Li)��, �����������L12�;���ṹ, �侧����[22]Ϊa=0.4010nm�� �ġ�(Al3Li)��ľ���ṹ��ͼ2(c)��ʾ��
2 ���㷽������
������ԭ�ӵļ۵��ӽṹ��������ָ�ù�����ԭ��������״̬�Լ�ԭ���γɹ��ۼ��ļ���ֲ��� ����EET[19]����, ԭ�ӵĹ��۵����Ƿֲ�����������ڡ� �ν���, �Լ�s����ԭ�ӵļ��ϡ� �����Ϲ��۵��Ӷ���(������n��)������ԭ�Ӽ��ʽ��ʾ:
![]()
ʽ�� RΪԭ�ӵ����뾶; �����µ���ֵ������[19]�е�ʽ(3)~(14)ȷ���� �����ڵĹ��۵���������дΪ
![]()
ʽ�� k1�� k2�ֱ�Ϊ������u�� vԭ�ӵĸ���; nuc�� nvc�ֱ�Ϊu�� vԭ�ӵĹ��۵�����; I��sΪn�������ĵ�ͬ����, ����ͬ������ѡȡ����������[19]�����ķ�����ȷ���� ���ڸ������Ľṹ��ȷ��, ʵ�龧[CM(22]����������[22]���Ѹ���, ���, ���ü�����[19]������ǿ��nA����, ���μ�����[20, 23-26]����ⲽ��, ����(1)�� (2)�ȷ�����, ��������������ԭ�ӳɼ��ļ۵��ӽṹ, ������BLD�о�ȷ��ԭ�ӵ��ӽ�״̬�� ����õ��ĸ������Ĺ��ۼ�������ڱ�1~6�� ���еĦҺ�K�ֱ��ʾԭ�ӵ��ӽ�״̬�Ϳ�λ��
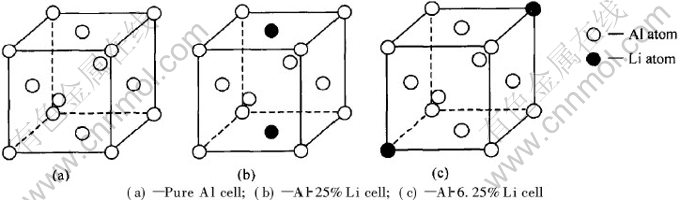
ͼ1 ��ͬLi������Al-Li����
Fig.1 Al-Li cell with different Li contents
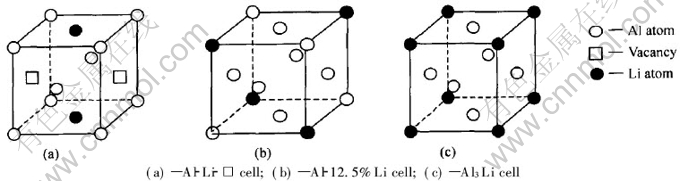
ͼ2 Al-Li-��λ�������������ṹ
Fig.2 FCC structures of Al-Li-vacancy cells
��1 ��Al�����Ĺ��ۼ�ǿ��
Table 1 Covalent bonds of pure Al cell

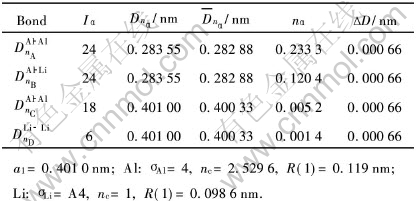
��2 Al-25%Li�����Ĺ��ۼ�ǿ��
Table 2 Covalent bonds of Al-25%Li cell
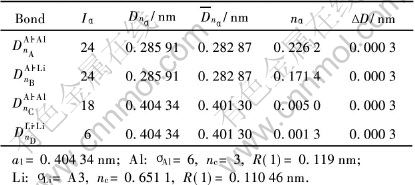
��3 Al-6.25%Li�����Ĺ��ۼ�ǿ��
Table 3 Covalent bonds of Al-6.25%Li cell
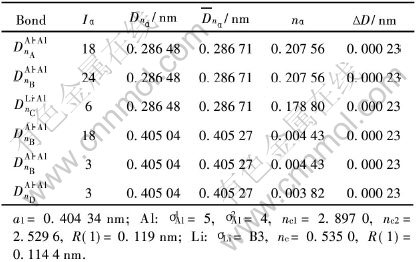
��4 Al-Li-�������Ĺ��ۼ�ǿ��
Table 4 Covalent bonds of Al-Li-�� cell
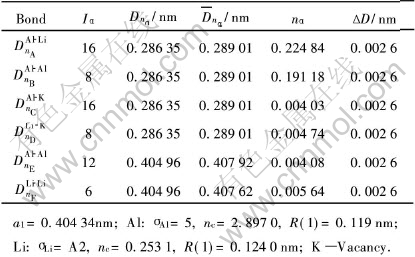
��5 Al-12.5%Li�����Ĺ��ۼ�ǿ��
Table 5 Covalent bonds of Al-12.5%Li cell
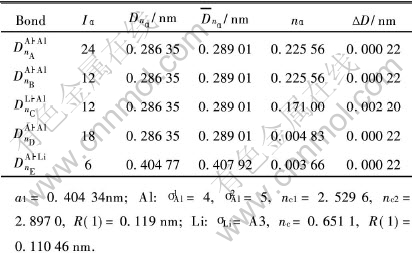
��6 Al3Li�λ�����Ĺ��ۼ�ǿ��
Table 6 Covalent bonds of Al3Li cell
3 ����������
�ɱ�2�� 3�� 5�ɼ�, �����3�ֲ�����λ��Al-Liƫ�۾����е���ǿ���ۼ���ΪAl��Al��, ��Li����Ϊ12.5%��25%�ľ�������Ǽ���ǿ��(nA=0.2250~0.2260)Ҫ�ȴ�Al�����ļ���Ǽ���ǿ��[23](nA=0.2086)Ҫǿ�� ����6.25%Li�ľ�������Ǽ����봿Al��������Ǽ��൱, ���ߵ���ǿAl��Al��nA�������, ԼΪ0.2075~0.2085�� �����Li�����ϵ�ʱ, Al-Liƫ�۾����ԺϽ������ǿ�����ԡ� �ɱ�4�ɼ�, ���ں���λ��Al-Li����, ��ǿ���ۼ��Ѳ�����Al��Al��, ����ת���Al��Li��, ����������������ڿ�λ���뾧����, ʹ��Al��Li�Ľ��������ǿ�� ʵ��[11, 12] �۲��Al-Li�Ͻ���̬���Ѵ���Al-Li�̳�����ṹ, ��Ҳ˵������Liԭ�����λ�н�ǿ�Ľ������, ��ʣ��λ�Ĵ����������γ�Al-Li�̳�����ṹ�� ����[2]ָ��Al-Li�Ͻ���̬�ĵ���ģ����Li�������Ӷ�����, ��������Al-Li-��λƫ�۾����Ĵ���, �ͺ�������һʵ�������к����Ľ��͡� ����Li����������, Liԭ�ӷ���Ŀ�λ��Ҳ����, ���, �������γɵ�Al-Li-��λ��ƫ�۾���������Ҳ����, ��ЩAl��Li����Ǽܽ�ǿ��ƫ�۾���, �����������̬�Ͻ�ĵ���ģ����
����ʱЧ�¶����ߺ�ʱЧʱ���ӳ�, �Ͻ��п�λ������, ����Al-Li-��λƫ�۾����п�λ�����ļ���Ǽ��۶Ͻ���, ����������ԭ�����顣 ��һ���̶�Ӧ������[6]������DSC����������, �����γɽṹ�����ȷ�ǰ����һ�����Ե����ȷ�, �������Ͽ��� �����Al-Li-��λƫ�۾����ܿ��ܾ�������[5, 6]ָ�����ںϽ���ʱ������Эͬ�����Spinodal�ֽ�IJ���, ��Щ����ṹ����ܿ��ܽ���Ϊ����[6]��ָ���Ĵ��ʱЧ�����γɦġ�(Al3Li)���ǰ�ṹ����̥�ṹ�� ����[20]����������Щƫ��ǰ�ṹ��Χ���ڽ϶�Ŀ�λ��, λ������ȱ�ݡ� ��Щ������ǰ��ƫ�۽ṹͨ���ǰ����п�λȱ�ݵĽṹ�� ���ڿ�λ�Ĵ���, ʹ�úϽ�����ԭ�Ӿ�����ɢ�������γɶ̳����ṹ��
����ʱЧʱ���һ���ӳ�, ���ȵ����������� �ɱ�6�ɼ�, �ġ����ǿ���ۼ�ΪAl��Al��, ǿ�ȴﵽnA=0.2333, ��Al-Li-��λƫ�۾�������ǿ����(nA=0.22484)�������, ����ǿ������, ��ΪnA=0.1244�� ��Ҳ�����ġ�(Al3Li)�ļ���ṹ������������, ��������[17]�����ļ���������һ�µġ� ���ڦġ�ļ���ǼܱȺϽ����ļ���Ǽܸ���ʵ, �������������нϺõĹ����ϵ�� ���, �����Ħġ������ܹ��ڹ���ǿ���Ļ�����������ǿ����Ͻ�ļ���ṹ, ��ߺϽ�ĵ���ģ��, ͬʱҲʹ�Ͻ��ǿ���нϴ����ߡ�
�ɱ�2�Ľ���ɼ�, ���ں�LiΪ25%�ľ���, Alԭ�ӵ�״̬������ߵĵ�6�ӽ�, �����3���۵��Ӷ�ת��Ϊ���۵���, ��������ɵ���, ��ʵ������ĵ����ʲ�û�������½�, �������ԭ��״̬�ľ���ʵ�ʳ��ֵĿ����Ժ�С�� ���������������, ˵�������̿�������Alԭ��״̬�����ܴ�ı仯, ��ԭ������ǺϽ��ڲ���ԭ������ú�ǿ, ��Ӧ���ܴ��Ե�ʡ� ͨ�������, Alԭ�Ӵ���������ȫ���۵���״̬�Dz��׳��ֵġ�
�ɱ�3�ͱ�5�ɼ�, ���ڲ�����λ��Al-Li������, Alԭ�ӵ��ӽ״��ڵ�4��5�ӽ�, Liԭ�Ӵ��ڵ�3�ӽס� ��LiΪ6.25%�ľ�����ǿ��Խ���, ���ܶԻ�����ǿ�����á� ��Li�����ϸߵ���12.5%�ľ���, �����ǿ, �ڴ��̬�Ͻ�������ǿ�����á�
�ɱ�4�Ľ�������Կ���, Al-Li-��λ������Liԭ�Ӵ��ڵ�2�ӽ�, �乲�۰뾶Ϊ0.1240nm, ��Alԭ�ӵĹ��۰뾶0.119nmҪ��, ��ǿ��ΪAl��Li��, ���������Al��Liԭ�ӵĵ縺�����ϴ�, ��λ�Ĵ���, ʹ��Liԭ�ӵ������չ, ����Alԭ��ת�Ƶ�ɵ�����, �����γ�Al-Liԭ��ƫ��״̬, �����ܴ������д�����Al-Li������չ���ơ� ���ڿ�λ���������Al��Li����Al��Al��Ҫǿ, ʹ�ô���λ��Al-Liƫ�۾����ڴ������м���������, �Ӷ��Դ��̬�Ͻ���һ����ǿ�����á� ��������λ��Al-Liƫ�۾����е�Liԭ�����ڵ�3�ӽ�, �乲�۰뾶Ϊ0.1144nm, ����Alԭ�ӵİ뾶0.119nmҪС�� ���ֲ�����λ�ľ�����Liԭ�����ڰ뾶��С, ʱЧ����������ͨ����λ��ɢ�ƶ��� ��Al-Li-��λ������, Liԭ�Ӱ뾶�ϴ�, ��Alԭ�ӵĹ��۽�Ͻ�ǿ, ������Լ����Ϊ����ԭ�Ӷ���ɢ�˶�, ����Ҫ�ϸߵ��¶Ȳ����ƻ�Al��Li���硣
���ϼ���ļ��ֿ��ܵľ����ṹ��, ���ܴ����������γɵ�ƫ�۾���ΪAl-12.5%Li��Al-Li-��λƫ�۾���, ���Ƕ����ܶԹ��ܴ��Ͻ���ǿ�����á�
4 ����
1) ����λ��Al-Li����������ǿ, ��ǿ���ۼ�ΪAl��Li���� �����е�Liԭ�Ӱ뾶��Alԭ�Ӱ뾶�������ڿ�λ����, ʹ��Liԭ���������˶���Χ������չ, ԭ�ӳɼ�������������ġ� Liԭ�Ӱ뾶�ϴ�ʹ��Liԭ����ɢǨ�ƽ��ѡ� Al-Li-��λƫ�۾����ܿ��ܾ��Ǵ��ʱЧ�����γɦġ�(Al3Li)���ǰ�ṹ����̥�ṹ��
2) ������λ�ľ���, ��ǿ��ΪAl��Al��, Liԭ�Ӱ뾶��С, �����ع�ʣ��λ�˶��� Li�����͵ľ�������ǿ�Ƚ���, �ԺϽ����ǿ�����ò����ԡ� ��12.5%Li��Al-Li����, �����ǿ, �ڴ��̬�Ͻ�������ǿ�����á� Li����Ϊ25%�ľ���, ����Ҫ��Alԭ�����3���۵��Ӷ�Ϊ���۵���, �������һ�����������ʵ��, ������ƫ�۾������ֵĿ����Ժ�С��
3) �ġ�(Al3Li)����������������, ����ǿ���ۼ���Al-Liƫ�۾�������ǿ���ۼ��ͻ���Ĺ��ۼ���Ҫǿ, ������ԺϽ������ǿ�����õ�����ԭ��
REFERENCES
[1]Lavernia E J, Srivatsan T S. Review of strength and fracture behavior and ductility of Al-Li alloys[J]. J Mater Sci, 1990, 25: 1137-1158.
[2]Noble B, Harris S J, Dinsdale K. The elastic modulus of Al-Li alloys[J]. J Mater Sci, 1982, 17: 461-468.
[3]Enrique J, Nicholas J. Review aluminum-lithium alloys[J]. J Mater Sci, 1987, 22: 1521-1529.
[4]Kassab T, Menand A, Chambreland S. The early stage of decomposition of Al-Li alloys[J]. Surface Science, 1992, 266: 333-336.
[5]Wei Y H, Wang S T. Experiment evidence for spinodal decomposition in Al-12.7%Li alloys[J]. Material Letter, 1996, 28: 123-127.
[6]Noble B. Evidence for pre �¡� precipitation events in an Al-Li alloys[J]. Scripta Metall, 1995, 33(1): 33-37.
[7]Meng F L, Chai Z G, Li J X. Small-angle X-ray scattering study of the �¡� phase in Al-Li alloys[J]. Mater Characterization, 2001, 47: 43-46.
[8]Noble B, Bray S E. The interfacial energy of �¡� precipitation in Al-Li alloy[J]. Mater Sci Eng A, 1999, A266: 80-85.
[9]Perez J I, Madariaga G. Quantitative analysis of �¡� precipitation kinetics in Al-Li alloys[J]. Acta Mater, 2000, 48(4): 1283-1296.
[10]Boukos N, Papastaikoudis C. The influence of �¡� precipitates on the electrical resistivity and low-field Hall coefficient of Al-Li alloys[J]. Phil Mag B, 1994, 70(1): 67-75.
[11]Ceresara S, Giarda A, Sanchez A. Annealing of vacancies and ageing in Al-Li alloys[J]. Phil Mag, 1977, 35(1): 97-110.
[12]Gregson P J, Flower H M. Role of vacancies in coprecipitation of �ġ�- and S-phase in Al-Li-Cu-Mg alloys[J]. Mater Sci Tech, 1986, 2: 349-353.
[13]Gaber A, Afify N. Characterization of the precipitates in Al-Li alloy using thermal measurements and TEM[J]. Physica B, 2002, 315(1): 1-6.
[14]Sato T, Tanhka N, Takahashi T. High resolution lattice images of ordered structure in Al-Li alloy[J]. Transactions of the JIM, 1988, 29(1): 17-25.
[15]Noble B, Bray S E. On the Al3Li metastable solvus in Al-Li alloy[J]. Acta Mater, 1998, 46(17): 6163-6171.
[16]Poduri R. Computer simulation of morphological evolution and coarsening kinetics of �ġ�(Al3Li) precipi-tation in Al-Li alloys[J]. Acta Mater, 1998, 46(11): 3915-3920.
[17]Guo X Q, Podloucky R, Xu J H. Cohesive and structural properties of Al3Li[J]. Phys Rev B, 1990, 46(18): 12432-12440.
[18]Pauling L. The Nature of the Chemical Bond[M]. San Simeon: Corrnell University Press, 1960. 300-400.
[19]������. ��������Ӿ����������[M]. ����: ���ֿ�ѧ����������, 1993. 1-90.
ZHANG Rui-lin. The Empirical Electron Theory of Solids and Molecules[M]. Changchun: Jilin Science and Technology Press, 1993. 1-90.
[20]��־��. �Ͻ�۵��ӽṹ��ɷַ���[M]. ����: ���ֿ�ѧ����������, 1990. 1-300.
LIU Zhi-lin. The Valence Electron Structure and Composition Design of Alloys[M]. Changchun: Jilin Science and Technology Press, 1990. 1-300.
[21]Li Z L, Ma C X, Liu Z L. Valence electron structure of high property steel and its composition design[J]. Acta Metallurgica Sinica, 1999, 12(4): 408-416.
[22]Mondolfo L F. Structure and Propertys of Aluminum Alloys[M]. London: Butterworths Press, 1976. 200-400.
[23]��Ӣ��, ������, ����. Al-Zn������۵��ӽṹ��Spinodal �ֽ�����[J]. �й���ɫ����ѧ��, 2004, 14(5): 730-735.
GAO Ying-jun, HAN Yong-jian, ZHAO Miao. Electron structure of Al-Zn solid solutions and spinodal decomposition[J]. The Chinese Journal of Nonferrous Metals, 2004, 14(5): 730-734.
[24]GAO Ying-jun, BAN Dong-mei, HAN Yong-jian. Atomic bonding and mechanical properties of Al-Mg-Zr-Sc alloy[J]. Trans Nonferrous Met Soc China, 2004, 14(5): 922-927.
[25]GAO Ying-jun, HAN Yong-jian. Electron structure and interface energy of GP zone in Al-Zn alloy[J]. Mater Sci Forum, 2005, 475-479: 3131-3135.
[26]��Ӣ��, ����ƽ, ����, ��. Al-Cu�Ͻ�������ļ۵��ӽṹ����[J]. ϡ�н���, 2003, 27(6): 845-849.
GAO Ying-jun, ZHONG Xia-ping, LIU Hui. Calculation on valence electron structures of metastable phase in Al-Cu alloy[J]. The Chinese Journal of Rare Metals, 2003, 27(6): 845-849.
������Ŀ: ������Ȼ��ѧ����������Ŀ(50061001); ������ѧ����������Ŀ(��ƻ�0342004-1); ������ʮ��ǧ�˲Ź��̡�������Ŀ(2001207)
�ո�����: 2004-11-29; ������: 2005-04-06
�����: ��Ӣ��(1962-), ��, ����, ��ʿ.
ͨѶ����: ��Ӣ��, ����; �绰: 0771-3236667; E-mail: Gaoyj@gxu.edu.cn
