
���±�ţ�1004-0609(2016)-05-1136-07
���۸���Һ��ֵĴ�����ʽ����Ե�Ƶ�Ӱ�����
�� ������ӿ���� �ڣ��� ־
(�й���ѧԺ ���̹����о��� ʪ��ұ����������������ҹ���ʵ����
��ɫ�����빤���ص�ʵ���ң����� 100190)
ժ Ҫ��
ͨ�����ۼ���͵��ʵ���о���Һ������ִ�����ʽ����Ը���Ƶ�Ӱ����ɡ������������ϼ�������98%�������Է�����ʽ��Cr(��)�����γ����۸����������������۸������ƽ���ͼ���֣������CrL3+��Cr(OH)L2+���ߵĵ绯ѧ���Թ����ڽϴ��ˮ����-���ĸ����Ӿ��롣ͨ���������۸����������Ũ�ȣ���������߸�������ʣ��ɸߴ�1.2 ��m /min�������������Ҫ��B(OH)3����ʽ���ڣ����pH���巶ΧΪ8~10����Al3+��ѵ�pH���巶ΧΪ3~3.5������0.6 mol/L Al3+ʹ���Ʋ��Ե�����ĺ��֮��(hcorner/hcenter)��11������2��������1 mol/L�����ʹhcorner/hcenter��5������3��Al3+���ƶƲ�����Ե����ø�Ϊ���ԡ�
�ؼ���: ��Һ��֣����۸���������������ʣ��Ʋ������
��ͼ����ţ�TQ151.8���� ���ױ�־�룺A
���۸�����������ܵ����ƣ����۸���ɫ��Ƽ������ܹ�ע��1854�꣬������������۸���Һ�е�������������о�[1]��Ȼ�����������۵�Ƽ����Ŀ��ٷ�չ��ֱ��1974�꣬�ſ���������ʵ�û���Alecra-3�Ȼ������۸���ƹ���[2]��1981�꣬Ӣ���о��߿��������������۸���ƹ���[3]����20���ͺ��ڣ����۸���ƹ��յõ�Ѹ�ٷ�չ��2003�꣬MacDermid��˾�Ƴ������������۸����ҺMacDermid��ʹ������ʵõ�������ߣ����������۸����װ���ԶƲ�Ļ���Ҫ��[4]����ƹ������Ѿ���ʼ��ҵ��Ӧ�á����۸���ϵ��ƺ�������Ͻ��Ը��ƶƲ��������ѧ����Ҳ�ܵ�Խ��Խ�㷺�Ĺ�ע[5-6]��
һ����Ϊ�ڼ����۸����Һ�У�Cr(��)������������λ������Cr(H2O)63+��ʽ���ڣ����ֻ�����ʮ���ȶ�������ֱ�ӵ绯ѧ��ԭΪ��������ͬʱ�����⸱��Ӧ��Ϊ���ң�ʹ��������OH-�����ۼ�����Cr(H2O)63+��Ӧ�����ȶ������Ż������ֹ��Cr(��)�Ļ�ԭ[7]��Ϊ��ʵ�����۸��ĵ绯ѧ��ԭ��ͨ����Ҫ�����۸���Һ������һ�������л����ᡢ�������л���λ�塣POLUKAROV��[8]��Ϊ�л����ӿ�ȡ��Cr(H2O)63+�е�ˮ���ӣ����ɻ��Ը�������E1-SHARIF��[9]����Ϊ�л�������谭���Ż�������γɣ��Ӷ��ٽ�Cr(��)�ĵ绹ԭ��ZENG��[10]֤ʵ�ڼ����۸���Һ�У����۸�������Cr(H2O)63+��ʽ���ڣ�ͨ������λ������Ϸ�Ӧ���ɾ��е绯ѧ���Ե�Cr(H2O)5L3+��HE��[11]������Һ����ϵ�о������۸��ĵ绯ѧ��ԭ��Ϊ��
���۸���Һ��Ҫ�����Ρ���ϼ���������������κ����Ӽ�����ɣ�������ϼ������Ӽ���ҪΪ�����л�С���ӻ�������¶�Һ�ɷֺ����ʸ���[11-12]����Һ��ּ��������̬����Ӱ���������������ɡ����۸���ƹ��պͶƲ�����[13-16]��Ȼ����Ŀǰ��û�жԵ�����������۸���Һ������ֵĴ�����ʽ�����������ʶ����������ϵͳ������Һ������������Ρ���ϼ�����������ڶ�Һ�еĴ�����ʽ�����о���Һ��ֶ����۸���Ƶ�Ӱ����ɡ�
1 ʵ��
���۸���Һ�ɷּ���1����Cr2(SO4)3��6H2OΪ��ѧ���⣬�����Լ���Ϊ����������Һ��ȥ����ˮ���ơ�
��Һ���Ʒ������ȳ�ȡ392 g������������Ϊ40%���������Һ������ȥ����ˮ����������48 g�壬��Ͼ��Ⱥ���ȥ����ˮ��500 mL������90 �������м���3 h����������Һ��ȴ�����º����μ�����ᡢ�����������ᡢ�����ƣ�����ȥ����ˮ��Ͼ��ȣ�Ȼ�����pH��������1 L�����ǰ����ҺpH���ڵ�1.5��
��1 ���۸���Һ�ɷ�
Table 1 Compositions of Cr(III) baths
���õ��۵�ƣ���������3 cm��3 cm��ҿ��Ϳ��缫����������4 cm��4 cm����ͭƬ��ͭ�������ȳ�������ȥ����ˮ��ϴ����ĥ�⣬Ȼ��绯ѧ�⣬ˮϴ��������á��ڵ��ǰ��ͭ����������10%(��������)�������5 min�����ú�����ƣ���ҺpHΪ1.5�������ܶ�Ϊ20 A/dm2���¶�Ϊ30 �档
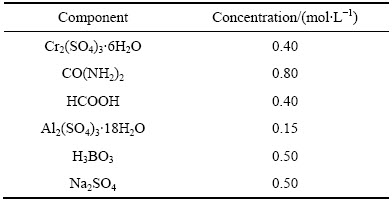
���Ʋ���Ȳ�������ET-2�����Զ�ܵ�����Dz��������ݷ����ڶ��ɣ��������������һ��ʱ���Ʋ����ɵ��ʱ����������Ʋ�ƽ������ó�ȡ��������������ȡ������[17]���Ƚ������жƲ�ĶƼ���ȥ����ˮ��ϴ�ɾ�������ȡ����������30%(��������)���Ὣ�Ƽ�����10 min��ʹ���Ʋ��ܽ⣬��ȥ����ˮ��ϴ�ɾ�����������������֮��Ϊ�Ʋ�����m(g)������ƽ���������ʰ���ʽ���㣺
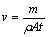 (1)
(1)
ʽ�У���Ϊ���Ʋ�������ʣ�cm/min����Ϊ���ܶȣ�7.19 g/cm3��AΪ���������cm2��tΪͨ��ʱ�䣬min��
2 ���������
2.1 �����ӵĴ�����ʽ
�ڵ����������Һ�У�Cr(��)�Զ��������ӵ���ʽ���ڣ���������ɫ����ɫ������ɫ��Cr(H2O)3+����ɫ��������������ӽ���Cr(H2O)3+�Ƚ�ʱ����Һת��Ϊ��ɫ���������ȡ����Խ����ɫԽ��[18]�����о�����ʹ�õ����������ɫ�ģ�Ӧ��һ������Crm(SO4)n(3m-2n)���ڡ�VINOKUROV��[19]�ܽ��������ˮ��Һ�п��ܴ��ڵ����۸�������Ӽ���Ӧ���ȶ��������������Һ�д��ڶ��ָ����������������ҺpH�йء���pHһ��ʱ�����ø�Ԫ���غ�(ʽ(2))����Ԫ��(ʽ(3))�ɼ��������ֵ�ƽ����ɡ�
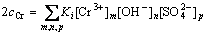 (2)
(2)
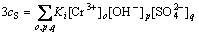 (3)
(3)
ʽ�У�cCr��cS�ֱ�Ϊ��Һ��CrԪ�غ�SԪ�ص�Ũ�ȣ�mol/L��m��n��l��o��p��qΪ������Ӽ�������صij�����KiΪ�ȶ�������
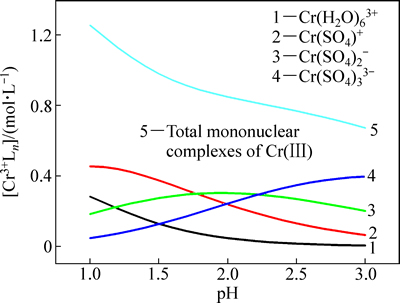
ͼ1 0.8 mol/L�����ˮ��Һ��pH�����۸���������������ʽ��Ӱ��
Fig. 1 Effects pH on form of trivalent chromium mononuclear complexes in 0.8 mol/L Cr2(SO4)3 aqueous solution
�������Ũ��Ϊ0.8 mol/Lʱ���������Һ����Ҫ�ĵ��˸�����P�ܵ��˸������Ũ�ȣ���ͼ1��ʾ�����о�����ʹ�õ������Ϊ��ɫ��Һ����pH����1.5ʱ��Cr(SO4)+��Cr(SO4)2-��Cr(SO4)33-��Ũ�ȶ�����Cr(H2O)63+��Ũ�ȣ�����������˵����������ӽ�����Cr(H2O)63+�Ƚ���Һ��Ϊ��ɫ����ϡ�����pH���ӣ�Cr(H2O)63+��Cr(SO4)+Ũ�ȼ��٣�Cr(SO4)33-Ũ�����ӣ���Cr(SO4)2-�������ټ��٣����ܵ��˸������Ũ�Ƚ��͡�ֵ��ע����ǣ�Cr(H2O)63+��Ũ��������ҺpH���߶����ͣ���pHֵΪ1.5ʱ����Ũ�Ƚ�Ϊ0.1 mol/L���ý�����ZENG��[10]����������������ʱ�����������Һ�и�����ȫ����Cr(H2O)63+��ʽ�������Բ�ͬ��
2.2 ��ϼ��Ĵ�����ʽ
���������۸���Һ��õ�һ����������ϼ�������ɷֽ�ΪHCOO-��H+������ˮ��Һ�����ϵ��ΪpKa=3.75����ˣ��ɼ����ˮ��Һ��pH�Լ���Ĵ�����ʽ��Ӱ�졣��ͼ2��ʾ���ڶ�Һ���ƺ͵��ʱ(pHΪ1.5)����������Һ����Ҫ��HCOOH���ӵ���ʽ���ڣ�ռ�ܼ����98%���ϡ�
����ˮ��Һ�е����ϵ��ΪpKa=0.18[20]���ݴ˿ɼ������ˮ��Һ��pH����Ĵ�����ʽ��Ӱ�졣��ͼ3��ʾ���ڶ�Һ���ƺ͵��ʱ(pH=1.5)��������Һ����Ҫ��CO(NH2)2���ӵ���ʽ���ڣ�ռ�����98%���ϡ�
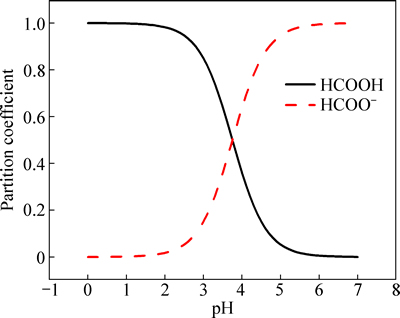
ͼ2 pH�Լ��������ʽ��Ӱ��
Fig. 2 Effect of pH on form of formic
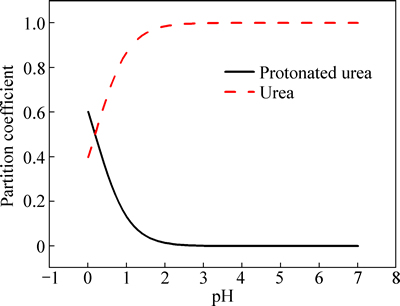
ͼ3 pH���������ʽ��Ӱ��
Fig. 3 Effect of pH on form of urea
����ͼ1�Ľ�������������Һ����۸��������ܹ��븺���Ե���������ӻ�ˮ�����γ����������������û�е绯ѧ���ԣ����Ե绯ѧ��ԭΪ������������IBRAHIM��[21]���о������������ͨ������CrLn3+����ʽ��ͼ2��3�Ľ���Ѿ�֤ʵ�����������Ϊ��ϼ����ڶ�Һ���ƺ͵�������£���Ҫ���Է��ӵ���ʽ���ڣ��ʵ����ԣ�Ҫʵ�����������������ɸ�������������ʵ����ϼ���������������ӻ�ˮ���Ӽ��ȡ����Ӧ���ӵ��Ժ�Cr(H2O)63+�Ľṹ�Ƕȣ�ȡ����Ӧ�Ķ���ѧ��������������ˣ����۸������������������ʽϵ͡��������ߵ�ǰ���о�����[22]��ͨ����߶�Һ�����¶����������۸��������л���������Ϸ�Ӧ����������۸������������������ʣ�����ZENG��[10]�Ľ�����ͬ����ˣ������۸���Һ���ƹ����У�������Ҫ�ϸߵ��¶Ⱥͽϳ���ʱ�䡣
2.3 ������Ĵ�����ʽ
���۸���ƹ������������������أ�����-��Һ����pH�ɴﵽ8���ϣ��������۸����������������������Ż���Ӧ�������ȶ������۸����Ӷ���Cr3+���ԴӾۺ������������������ԭ��Ϊ�˷�ֹ���Ż���Ӧ�������۸����������ͣ������ֶ�ҺpH�ȶ�����Һ��ͨ����Ҫ���뻺������ڵ�������У������dz����Ļ��������ΪһԪ�ᣬ��ˮ�з������·�Ӧ��
B(OH)3+OH- [B(OH)4]- (4)
[B(OH)4]- (4)
�÷�ӦpKa=9.24����ͼ4(a)��ʾ���ڶ�Һ���Ʒ�Χ��(��2)��������Ҫ��B(OH)3����ʽ���ڡ���pHֵ����ʱ��B(OH)3������Һ�е�OH-��Ӧ����[B(OH)4]-�������Ч��������ѻ��巶ΧΪ8~10֮��(��ͼ4(b))��
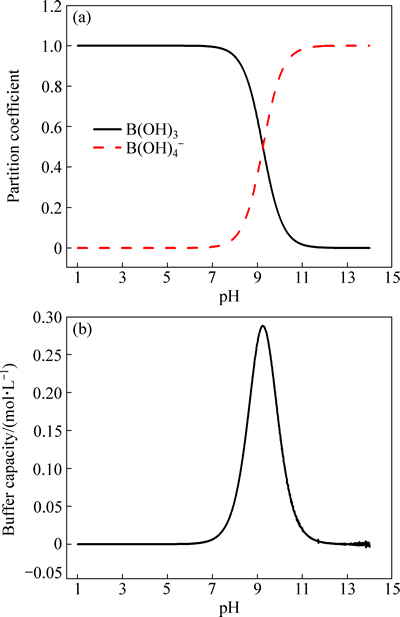
ͼ4 pH������Ĵ�����ʽ�����Ỻ��������Ӱ��
Fig. 4 Effect of pH on form of boric acid (a) and buffer capacity of boric acid (b)
������������һ�ֳ��õĻ������������[23]��֪��Al3+��ˮ��Һ�н�����ˮ�ⷴӦ�����ɸ�����ʽ������ˮ�����ӡ��ڶ�Һ���������£�Al3+�ڲ�ͬpH�µ�Ũ����ͼ5(a)��ʾ����ͼ5(a)��֪����pH��3���ӵ�3.5ʱ��Al3+Ũ�ȴ�0.28 mol/L���罵��0.01 mol/L����˵���ڸ�pH�����£�Al3+���Һ�е�OH-������Ӧ�����ĵ������⸱��Ӧ�����ɵ�OH-������Ч�������۸������������OH-��Ӧ�������۸����Ӷ�����ͼ5(b)��ʾ��Al3+��ѻ��巶Χ��3~3.5�䣬��ͼ5(a)�Ľ����һ�¡�
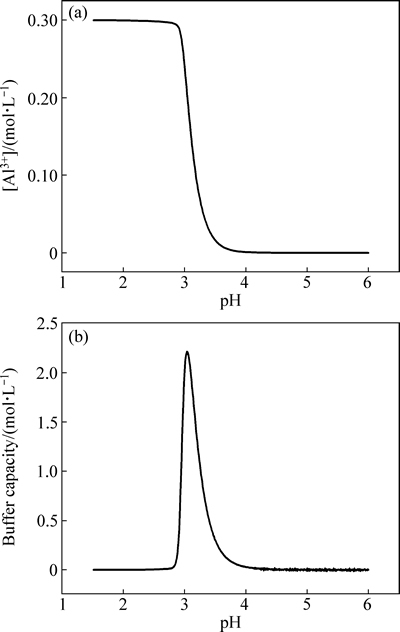
ͼ5 ��ҺpH��������Ũ�ȵ�Ӱ��������ӵĻ�������
Fig. 5 Effect of pH on Al3+concentration (a) and buffer capacity of aluminum ion (b)
�����۸���Һ��ͬʱ������������������������߸��Ե���ѻ��巶Χ��ʹ��Һ�Ļ��������õ���ߣ�����Ч�������۸���ƹ���pH���ر��ǵ缫/��Һ���洦pH�Ŀ������ߡ�
2.4 ���۸�����������ȷ��
����ǰ�ڵ��о���֪[20]�����۸����������Cr(OH)L+(LΪ�����)�����۸��绹ԭΪ���������м�����CrL3+����һ������ֱ�ӵ绹ԭ���ɡ�ͨ����Һ���䣬����ֱ�ӻ�ñ�CrL3+�绯ѧ���Ը��ߵ�Cr(OH)L2+�������ƹ���CrL2+��OH-�Ļ���ȡ����Ӧ������������۸�������ʡ�
ZENG��[24]��Ϊ������λ�����۸��������ֻ�е�������������ļ�����2.04 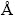 ʱ��������е�ˮ���Ӳ������ѳ�����õ����۸������Ӳ��п����ڵ缫���滹ԭ����ˣ�����ͨ������������ֶ�ȷ�����۸�������ӵķ��ӽṹ���Ӷ��жϸ������ĵ绯ѧ���ԡ�
ʱ��������е�ˮ���Ӳ������ѳ�����õ����۸������Ӳ��п����ڵ缫���滹ԭ����ˣ�����ͨ������������ֶ�ȷ�����۸�������ӵķ��ӽṹ���Ӷ��жϸ������ĵ绯ѧ���ԡ�
�����ܶȷ������۵Ĺ����ݶȽ����㷨������Cr(H2O)63+��CrL3+��Cr(OH)L2+��ƽ��͡�ͼ6��ƽ���1�е�Cr3+��H2O���Ϊ2.02 ���������������ǿ��ˮ���ӣ���ˣ�Cr(H2O)63+�е�һ��ˮ�������ױ���ȡ�����ɾ��е���Ե�Cr(H2O)5L3+����ʾΪCrL3+������Ľ����ʹCr3+��H2O֮��ľ���������ͼ6��ƽ���2��ʾ���������ˮ���������ĸ����ӵľ���Ϊ2.07 �����ԣ����ˮ���������������ĸ����ӣ���ʱCrL3+�����������Ϻ��������绹ԭ������ҺpH��1.4~2����ʱ������pH�����ӣ���Һ��OH-��֮���ӡ�����ǰ���о�����[21]������ҺpH���ӵ�1.83ʱ������Ƶĵ���Ч�ʿɴﵽ28.9%����ƽ����Ϸ������������������CrL3+�ϲ���һ��OH-��������γ�����ƽ���3��4�����У�ƽ���3��ˮ���������ĸ����ӵļ��ɸߴ�2.15 ����ƽ���4��ˮ���������ĸ����ӵļ���2.08 ������Ȼ��ƽ���3�е�ˮ����Ҫ��CrL3+�е�ˮ���Ӹ������ѳ�����ˣ������ж�Cr(OH)L2+�DZ�CrL3+�ĵ绯ѧ���Ը��ߵ���������pH����ʱ������Ƶĵ���Ч�ʻ����ӡ�
ͼ6 �������۸�������ƽ���
Fig. 6 Equilibrium geometric structures of typical Cr(III) complexes
2.5 ���۸�������������Ը�������ʵ�Ӱ��
��Һ�е����۸��������������Ƶ�ֻ�����۸����������[24]��ͼ7(a)Ϊ���Ƶ������-����Һ��ϡ�͵���ͬ������Ȼ��������������֣������õ��Ķ�Һ�Ļ��������Ũ�������������-���Ũ�ȳ����ȣ��ʿ��ü�������Ũ������ӱ�ʾ���۸���������ͼ7(b)�Ķ�Һ�������Ũ��Ϊ0.4 mol/L����ϼ����Ũ��Ϊ0.1~0.5 mol/Lʱ��������������Ͳ�ͬŨ����Ļ����ҺȻ��������������֡�ͼ7(a)��ʾΪ���۸����������Ũ�����������ʵĹ�ϵ����ͼ����ʾ��Ũ�ȷ�Χ�ڣ�����������������۸����������Ũ�ȵ����߶�Ѹ�����ߣ����ǵ����۸�����������Ũ�����ӵ�һ��Ũ��ʱ����Ը�������ʵ�Ӱ�����������۸��������ʸߴ�1.2 ��m/min��
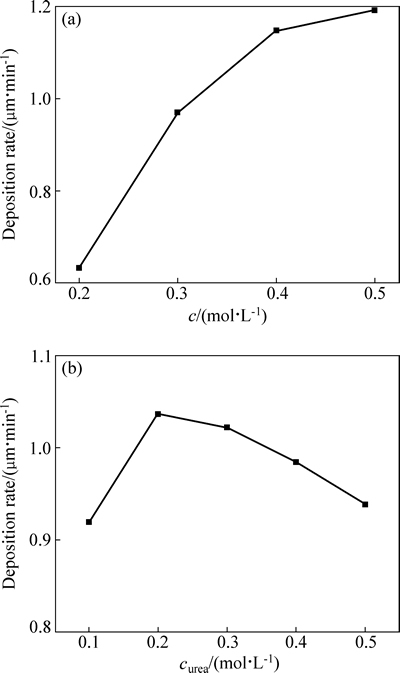
ͼ7 ���۸����������Ũ�Ⱥ���Ũ�����������ʵĹ�ϵ
Fig. 7 Relationships between concentration of Cr(��) active complexes (a) and concentration of urea (b) and deposition rate
ͼ7(b)��ʾΪ��Һ����Ũ�����������ʵĹ�ϵ����ͼ7(b)�п�֪������Ũ��С��0.2 mol/Lʱ�����������������Ũ�ȵ����Ӷ����ӡ�������Ũ�ȼ�������ʱ����������ʷ������½���ʵ�������õ����۸����ӵ�Ũ��Ϊ0.8 mol/L������������ʴﵽ���ֵʱ���õ���Ũ��ֻ��0.2 mol/L����˵�������Ƕ�Һ�����е����۸����Ӷ������γɻ�����������Ũ�ȼ�������ʱ����Һ�е���������۸�����������һ����Ӧ���ɻ��Խϵ͵�CrLn3+(n��2)���Ӷ���ɸ�������ʵ��½�[10]��
2.6 ������ԶƲ�����Ե�Ӱ��
���ݻ����������ԭ�������ӻ������Ϊ�����վ������⸱��Ӧ�ͷų���OH-���Ӷ�������������Cr(OH)3�����۸����Ӷ��������ɣ���������۸����ӵĵ�������ʡ���������������۸������������Ӱ�죬���ԶƲ�ľ�������Ӱ�졣Ϊ�˱����Ʋ�ľ����ԣ����õ�����Dz�ȡ�Ʋ����ĺ��hcenter�ͶƲ����½Ǻ��hcorner�����������߱�ֵhcorner/hcenter����ֵԽ�Ʋ�Խ�����ȡ����ڳ��ε����ֲ���ԭ�߽Ǵ��ĵ����ܶ�Ҫ�������Ĵ��ĵ����ܶȣ���ˣ�����-��Һ�����pH����ֱ�Ե������С���ص㡣�ڽϵ͵�pHֵ��Χ�£�pH����ٽ����ij�������������ļ��������pH�����ӣ������ǶԱ߽Ǵ���Ӱ��Ч��������ˣ�������ļ����ʹ�Ʋ��ȷֲ����ȡ�
��ͼ8��֪��Al3+Ũ�ȴ�0���ӵ�0.6 mol/Lʱ��hcorner/hcenter��11������2���仯��Լ5������H3BO3��0���ӵ�1.0 mol/Lʱ��hcorner/hcenter��5������3��ֻ�仯��Լ2�����ɴ˿ɼ���Al3+�ԶƲ�����Ե�Ӱ�������������Al3+����ѻ��巶Χ��3~3.5����ͼ5(b)��ʾ����ͼ4(b)��ʾ�����8~10�ķ�Χ��ƫ���ԣ���Al3+�Ļ��巶Χ�������۸���Һ��pHֵ��Χ��
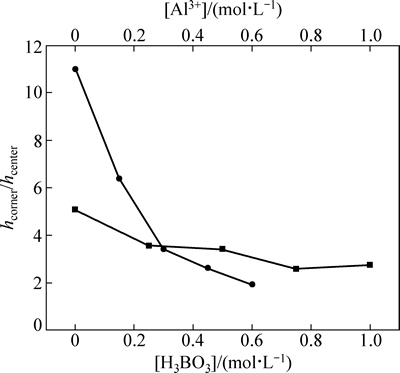
ͼ8 ����Ũ�ȡ�������Ũ�ȷֱ���hcorner/hcenter��ϵ
Fig. 8 Relationship between of hcorner/hcenter and concentration of boric acid and aluminumion
3 ����
1) ͨ�������۸���Һ������ִ�����ʽ�ķ����������������Һ��Cr(��)��Ҫ�Ե��˸��������ʽ���ڣ���ϼ��������98%�������Է�����ʽ���ڣ�����Cr(��)��ϣ��������۸���������
2) Cr(OH)L2+�������ˮ���������ĸ����ӵļ�����Դ���CrL3+�ģ���ˣ�Cr(OH)L2+���и��ߵĵ绯ѧ���ԡ�
3) ����������������۸�������Ũ�ȵ����Ӷ����ӣ�������߿ɴ�1.2 ��m/min��
4) ����ҺpHΪ1.5ʱ������Al3+ʹhcorner/hcenter��ֵ����Լ5����H3BO3ʹ�ñ�ֵ����Լ2����˵��Al3+�ԶƲ�����Եĸ������ø����ԡ�
REFERENCES
[1] ����ƽ, ��ҫ��, ղ����. ���۸���Ƶ��о��뷢չ[J]. ���漼��, 2003, 32(3): 5-7.
LIU Jian-ping, HU Yao-hong, ZHAN Yi-teng. Research and development of trivalent chromium plating[J]. Surface Technology, 2003, 32(3): 5-7.
[2] ������, ��ۼ��. ��������ϵ���۸����װ�θ���ͻ��[J]. Ϳװ����, 2011, 4: 3-9.
GUO Cong-wu, LAI Huan-wen. Breakthrough in decorative trivalent chromium plating of sulphate system[[J]. Coating and Plating, 2011, 4: 3-9.
[3] ������, �� ��, ������. ���۸������ε�Ƹ��ķ�չ��״[J]. ����뾫��, 2009, 31(1): 13-17.
LI Yong-yan, LI Ning, TU Zhen-mi. Development status of chromium plating from trivalent chromium sulfate solution[J]. Plating and Finishing, 2009, 31(1): 13-17.
[4] �ŵ�ѧ, ������, �� ��, ������, ��־��. ���۸���Ƶ��о���״����չ[J]. ���ϱ���, 2010, 43(4): 29-34.
DU Deng-xue, SUI Yong-hong, ZHOU Lei, LI Wen-peng, ZHANG Zhi-peng. Current status of research and development of trivalent chromium plating[J]. Journal of Materials Protection, 2010, 43(4): 29-34.
[5] MAHDAVI S, ALLAHKARAM S R. Composition, characteristics and tribological behavior of Cr, Co-Cr and Co-Cr/TiO2 nano-composite coatings electrodeposited from trivalent chromium based baths[J]. Journal of Alloys and Compounds, 2015, 635: 150-157.
[6] RAMEZANI-VARZANEH H A, ALLAHKARAM S R, ISAKHANI-ZAKARIA M. Effects of phosphorus content on corrosion behavior of trivalent chromium coatings in 3.5% wt.% NaCl solution[J]. Surface and Coatings Technology, 2014, 244: 158-165.
[7] TU Z, YANG Z, ZHANG J. Cathode polarization in trivalent chromium plating[J]. Plating and Surface Finishing, 1993, 80(11): 79-82.
[8] POLUKAROV Y M, SAFONOV V A, EDIFARYAN A A, VYKHODTSEVA L N. Chrome plating from sulfate-oxalate Cr(III) baths. structure, composition, and corrosion behavior[J]. Protection of Metals and Physical Chemistry of Surfaces, 2001, 37(5): 447-451.
[9] EL-SHARIF M, MA S, CHISHOLM C U. Environmentally acceptable process for electrodeposition of hard chromium from chromium(��) electrolyte[J]. Transactions of the Institute of Metal Finishing, 1999, 77(4): 139-144.
[10] ZENG Z X, ZHANG Y X, ZHAO W J, ZHANG J Y. Role of complexing ligands in trivalent chromium electrodeposition[J]. Surface and Coatings Technology, 2011, 205(20): 4771-4775.
[11] HE X K, HOU B L, LI C, ZHU Q Y, JIANG Y M, WU L Y. Electrochemical mechanism of trivalent chromium reduction in 1-butyl-3-methylimidazolium bromide ionic liquid[J]. Electrochimica Acta, 2014, 130: 245-252.
[12] WIJENBERG J H O J, STEEGH M, AAENTS M P, LAMMERS K R, MOL J M C. Electrodeposition of mixed chromium metal-carbide-oxide coatings from a trivalent chromium-formate electrolyte without a buffering agent[J]. Electrochimica Acta, 2015, 173: 819-826.
[13] ���¿�, �°���, �����, ��С��, ��ȫ��. ���۸�������������Ni-Cr�Ͻ���[J]. �й���ɫ����ѧ��, 2006, 16(7): 1281-1287.
HE Xin-kuai, CHEN Bai-zhen, WU Lu-ye, LI Xiao-dong, HE Quan-guo. Process of pulse electrodeposition of nanocrystalline Ni-Cr alloy from trivalent chromium bath[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(7): 1281-1287.
[14] CHIEN C W, LIU C L, CHEN F J, LIN K H, LIN C S. Microstructure and properties of carbon-sulfur-containing chromium deposits electroplated in trivalent chromium baths with thiosalicylic acid[J]. Electrochimica Acta, 2012, 72: 74-80.
[15] LEE J Y, KIM M, KWON S C. Effect of polyethylene glycol on electrochemically deposited trivalent chromium layers[J]. Transactions of Nonferrous Metals Society of China, 2009, 19: 819-823.
[16] �����, ���Ӻ�. �����������۸��������Ϊ��Ӱ��[J]. ���ϱ���, 2006, 39(7): 26-28.
WU Hui-min, AI You-hong. Effect of brightener on electrodeposition of trivalent chromium in sulfate bath[J]. Journal of Materials Protection, 2006, 39(7): 26-28.
[17] MCDOUGALL J, EL-SHARIF M, MA S. Chromium electrodeposition using a chromium (III) glycine complex[J]. Journal of Applied Electrochemistry, 1998, 28(9): 929-934.
[18] �� ��, �� ��. ��������������Ӧ��[M]. ����: ��ѧ��ҵ������, 2003.
DING Yi, JI Zhu. Production and application of chromium compound[M]. Beijing: Chemical Industry Press, 2003.
[19] VINOKUROV E G, KUZNETSOV V V, BONDAR V V. Aqueous solutions of Cr(��) sulfate: modeling of equilibrium composition and physicochemical properties[J]. Russian Journal of Coordination Chemistry, 2004, 30(7): 496-504.
[20] MOHAN S, VIJAYAKUMAR J, SARAYANAN G. Influence of CH3SO3H and AlCl3 in direct and pulse current electrodeposition of trivalent chromium[J]. Surface Engineering, 2009, 25(8): 570-576.
[21] IBRAHIM S K, WATSON A, GAWNE D T. The role of formic acid and methanol on speciation rate and quality in the electrodeposition of chromium from trivalent electrolytes[J]. Transactions of the Institute of Metal Finishing, 1997, 75(5): 181-188.
[22] LI L, WANG Z, WANG M Y, ZHANG Y. The enhancement of Cr(III)electrodeposition by temperature and pH optimization during bath preparation[J]. International Journal of Minerals Metallurgy and Materials, 2013, 20(9): 902-908.
[23] ������. ����ˮ��ѧ��Ӧ������̬���[J]. ������ѧ�Կ�, 1987, 8(2): 1-9.
LUAN Zhao-kun. Chemical reaction of aluminum and its existing form[J]. Journal of Environmental Science, 1987, 8(2): 1-9.
[24] ZENG Z X, SUN Y L, ZHANG J Y. The electrochemical reduction mechanism of trivalent chromium in the presence of formic acid[J]. Electrochemistry Communications, 2009, 11: 331-334.
Existing forms of key components in trivalent chromium plating solution and its effects on chromium plating
LIU Yang, WANG Ming-yong, LI Lei, WANG Zhi
(Key Laboratory of Green Process and Engineering,
National Engineering Laboratory for Hydrometallurgical Cleaner Production Technology,
Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China)
Abstract: The components in trivalent chromium plating solution are complex. The existing forms of key components in trivalent chromium solution and their effect on chromium electrodeposition were studied by theoretical calculations and plating experiments. The results indicate that 98% of both urea and formic acid in the molecule form are combined with Cr(��) to form active Cr(��) complexes. According to the equilibrium conformation diagram of Cr(��) complexes, higher electrochemical activity of Cr(OH)L2+ than CrL3+ is ascribed to the larger distance between water molecules and chromium ion. The electrodeposition rate of Cr is enhanced by increasing the concentration of active Cr(��) complexes. The maximum rate reaches up to 1.2 ��m/min. Boric acid existed in the form of B(OH)3, and the optimum pH buffer range is 8-10. For Al3+, the optimum pH buffer range are 3~3.5. When 0.6 mol/L Al3+ is added, the value of the hcorner/hcenter is decreases from 11 to 2. In the presence of 1 mol/L boric acid, the value decreases from 5 to 3. This means that the effect of Al3+ on the improvement of coating uniformity is greater.
Key words: bath component; active Cr(��) complexes; electrodeposition rate; coating uniformity
Foundation item: Project (2013CB632606) supported by the National Basic Research and Development Program of China; Project (51274180) supported by the National Natural Science Foundation of China; Project (2015036) supported by the Youth Innovation Promotion Association, CAS
Received date: 2015-06-17; Accepted date: 2016-01-04
Corresponding author: WANG Ming-yong; Tel: +86-10-82544818; E-mail: mywang@ipe.ac.cn
(�༭ ����)
������Ŀ�������ص�����о���չ�ƻ�������Ŀ(2013CB632606)��������Ȼ��ѧ����������Ŀ(51274180)���й���ѧԺ���괴�´ٽ���������Ŀ(2015036)
�ո����ڣ�2015-06-17�������ڣ�2016-01-04
ͨ�����ߣ�����ӿ�����о�Ա����ʿ���绰��010-82544818��E-mail��mywang@ipe.ac.cn
ժ Ҫ��ͨ�����ۼ���͵��ʵ���о���Һ������ִ�����ʽ����Ը���Ƶ�Ӱ����ɡ������������ϼ�������98%�������Է�����ʽ��Cr(��)�����γ����۸����������������۸������ƽ���ͼ���֣������CrL3+��Cr(OH)L2+���ߵĵ绯ѧ���Թ����ڽϴ��ˮ����-���ĸ����Ӿ��롣ͨ���������۸����������Ũ�ȣ���������߸�������ʣ��ɸߴ�1.2 ��m /min�������������Ҫ��B(OH)3����ʽ���ڣ����pH���巶ΧΪ8~10����Al3+��ѵ�pH���巶ΧΪ3~3.5������0.6 mol/L Al3+ʹ���Ʋ��Ե�����ĺ��֮��(hcorner/hcenter)��11������2��������1 mol/L�����ʹhcorner/hcenter��5������3��Al3+���ƶƲ�����Ե����ø�Ϊ���ԡ�
[1] ����ƽ, ��ҫ��, ղ����. ���۸���Ƶ��о��뷢չ[J]. ���漼��, 2003, 32(3): 5-7.
[2] ������, ��ۼ��. ��������ϵ���۸����װ�θ���ͻ��[J]. Ϳװ����, 2011, 4: 3-9.
[3] ������, �� ��, ������. ���۸������ε�Ƹ��ķ�չ��״[J]. ����뾫��, 2009, 31(1): 13-17.
[4] �ŵ�ѧ, ������, �� ��, ������, ��־��. ���۸���Ƶ��о���״����չ[J]. ���ϱ���, 2010, 43(4): 29-34.
[16] �����, ���Ӻ�. �����������۸��������Ϊ��Ӱ��[J]. ���ϱ���, 2006, 39(7): 26-28.
[18] �� ��, �� ��. ��������������Ӧ��[M]. ����: ��ѧ��ҵ������, 2003.
[23] ������. ����ˮ��ѧ��Ӧ������̬���[J]. ������ѧ�Կ�, 1987, 8(2): 1-9.
