
���±�ţ�1004-0609(2012)08-2317-09
��þ����LiFePO4�ĵ��ӽṹ
�Ŷ���1, 2��������1�����껪2�����ı�1���� ��1��������1
(1. ���ڴ�ѧ ��ѧ�뻯��ѧԺ������ 518060��
2. ��������ҵ��ѧ�����о���Ժ ���Ͽ�ѧ�빤��ѧԺ������ 518055)
ժ Ҫ��
���û����ܶȷ������۵ĵ�һ��ԭ���о�����ӵ����������LiFePO4����Ni��Mg�ĵ��ӽṹ�����������Ni������ĵ��ӽṹ������������d����е���Ӱ�죬���ڲ��Ӹ���ʱ���ṹ�ȶ�����϶����ʹ���ӵ絼�����ӣ���ŵ�������ߣ�Mg���Ӹ���ʱ����������Liλʱ����϶�����ܾ����ͣ�ͬʱLi���ӵ���ɢ�˶�Ҳ��ߣ������ڸ��ԣ�����Feλ����ʱ�������渽��Fe-d����е���Ӱ���ܴ���ʹ��϶���ӣ������ڵ��ӵ絼�����IJ��ӷ���Ҳ����Li��O��������ǿ��������������ɢ���������ڸ��ԡ���ˣ�ͨ�����ӿɶ�LiFePO4���۽ṹ����Ӱ�죬�Ӷ�Ӱ����绯ѧ���ܣ��������۽ṹ��Ҫ��d����е����˶��Է����渽���ܴ���Ӱ�죬����������ӵĻ��ϼۺͰ뾶�ء�
�ؼ��ʣ�
LiFePO4����������þ���������ӽṹ����һ��ԭ����
��ͼ����ţ�O64 ���� ���ױ�־�룺A
Electronic structure of LiFePO4 doped with Ni and Mg
ZHANG Dong-yun1, 2, ZHANG Pei-xin1, SONG Shen-hua2, HUI Wen-bin1, HUANG Lei1, REN Xiang-zhong1
(1. College of Chemistry and Chemical Engineering, Shenzhen University, Shenzhen 518060, China;
2. School of Materials Science and Engineering, Harbin Institute of Technology Shenzhen Graduate School, Shenzhen 518055, China)
Abstract: LiFePO4 doped with Ni and Mg was simulated by using the first-principles. Through the energy band, PDOS and population analysis, it shows that, when the transition metal element Ni is used as dopant, the structure is becomes stable and the band gap reduces when Ni is doped on either Li(M1) or Fe(M2) sites, which results in the increase of the electronic conductivity and the discharge-recharge rate, because the electronic structure and energy properties of the transition metal oxides are greatly affected by the electrons in orbital d. When non-transition metal element Mg is doped at Li-site, both the band gap and total energy decrease, and the lithium ion diffusivity improves. When Mg is doped at Fe-site, the band gap increases because of the influence of electronics in Fe-d orbitals near the Fermi level, which is not beneficial to the electronic conductivity. The covalent interaction between Li and O strengthens through the analysis of population, which is not benefit for the lithium ion diffusivity and modification. So, the electrochemical properties of LiFePO4 are influenced by the changes of microstructure which are affected by dopants. The electrons in orbital d mainly affect the electronic structure near the Fermi level, while has no relative with the chemical valence and atomic radius of dopant.
Key words: LiFePO4; Ni-doping; Mg-doping; electronic structures; first principles
��1997��PADHI��[1]�״α������ʯ�ṹ��LiFePO4����������ӵ����������������������нϸߵ�����������������ѭ�����ܡ����õ����ȶ��ԡ���Դ�ḻ���۸�����������Ѻã��������Ϊ����һ������ӵ�أ��ر�������Ӷ�����غʹ��ܵ�ص���ѡ�������ϣ����ϵ͵ĵ��Ӽ����Ӵ������谭�˸ò��ϵ�ʵ��Ӧ�á�
CHUNG��[2]�о����֣����ý���������(Mg��Zr��Al��Nb��)���Ӻϳɾ���������ȱ�ݵ�LiFePO4������˵��ӵĵ����ʣ���绯ѧ����Ҳ�õ����Ը��ơ�NAKAMURA��[3-4]��ZHOU��[5-6]��MORGAN��[7]��WANG��[8]���߽����[9-10]Ҳ�������Ӳ��Ӻ���ϵĵ��ӵ絼�͵������кܴ����ߡ����Ƕ������Ӳ�����߲��ϵ绯ѧ���ܵ�ԭ��ȴһֱ���ڸ������飬������Ӳ��ӶԲ��ϵ绯ѧ���ܵ�Ӱ�����ȱ������ϵͳ���о�����û�й��ϵķ��������ۣ����Ӳ���������Ȼ����̽����[8-13]��
CEDER��[5-7, 14-16]��1998���״β��õ�һ��ԭ��������ָ������ӵ���������ϵĺϳɣ�������LiAlyM1-yO2(M=Co, Mn)��ϵ���Խ������ӵ����ʵ��Ӧ���������İ�ȫ�ԡ����ӵ絼�ʵ����⣬������Ҫ��ָ�����塣�˻�����[17-18]���û����ܶȷ������۵ĵ�һԭ�����о���LiMn2O4��LiNiO2�����ӻ�����Ľṹ���ȶ��ԡ�OUYANG��[19-20]ͨ����һ��ԭ�������о����������LiFePO4�е���ɢ������������LiFePO4������Liλ����Cr���ӵĵ��ӵ絼�ʵõ��������߶���绯ѧ����ȴû�����Ը��Ƶ�ԭ��WANG��[21]���õ�һ��ԭ��������Mo����LiFePO4�ĵ���̬�ܶȣ�����Mo�IJ��Ӹı���LiFePO4�����渽���ĵ���̬�ܶȵķֲ���������LiFePO4�ĵ��ӵ������ܣ�ģ������XASʵ����ԵĽ����һ�¡�ZHOU��[5]��LiFePO4�Ĵ�϶����UV-Vis-NIR����������VASP�����Ϸֱ����GGA ��GGA+U����������LiFePO4��LiMn2PO4 �ĵ��ӽṹ��
���ͱ���������������ϣ���δ�����й�þ(Mg)����(Ni)���Ӳ��ӵ�ģ����㣬Ϊ���۲����̽�����Ӳ��Ӹ���LiFePO4�绯ѧ���ܵ�Ӱ����������û��ƣ��������߲��û����ܶȷ������۵ĵ�һ��ԭ���о���Ni��Mg���Ӷ�LiFePO4���ӽṹ��Ӱ�죬������������Ӱ��LiFePO4�۵��ӽṹ�ı��ʡ�
1 ���㷽����ģ��
�ڱ�ģ���У����ϵ��ӽṹ�ļ���ģ����Ҫ����CASTEP�����������У�ģ�����ʱ��ͨ���Գ���(112)�е�Li��Fe��Ni��Mg�����û����õ��������Ӳ����ڲ�ͬλ�õ�LiFePO4ģ��(��ͼ1)��LiFe7/8M1/8PO4��Li7/8M1/8FePO4��M=Ni, Mg��
LiFePO4Ϊ���ʯ�ͽṹ������������ϵ[22]���ռ�ȺΪPnma��Z=4�������������£�a=1.032 9 nm��b=0.601 1 nm��c=0.469 9 nm���������Ϊ0.291 03 nm3��ͼ����ԭ�ӳ�Ť���������ܶѻ��ṹ��ÿһ����ԭ�����ڽ���4����ԭ�ӳɼ��γ�PO4�����壬��ռ���������4cλ��ÿһ����ԭ�Ӻ��ԭ�ӷֱ����ڽ���6����ԭ�ӳɼ��γ�FeO6��LiO6�����壬��ռ�ݰ������4c��4aλ[23]����b�᷽��ĽǶ���������bc���ϣ�FeO6������ͨ���������ӡ�LiO6��������b�᷽������ߣ��γ���״��һ��FeO6������ֱ���һ��PO4�����������LiO6�����干�ߣ�ͬʱ��һ��PO4�����廹������LiO6�����干�ߡ�LiFePO4����������Feԭ�Ӻ�һ��Pԭ�ӹ���һ��Oԭ�ӡ�
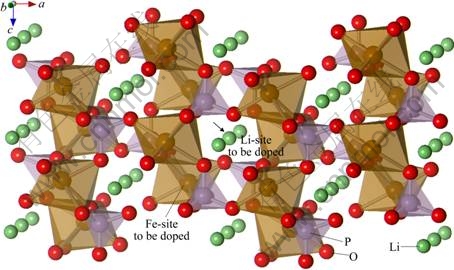
ͼ1 LiFePO4�����ʯ�ṹ
Fig. 1 Structure of LiFePO4
���������������ù����ݶȽ���(GGA)�µ�PBE�ݶ���������������ʱ��ԭ������ѡȡ���ǹ淶-�غ����ƣ�����Ԫ��ȫ���ó������������۵��Ӻ�ԭ��ʵ������á�Brillouin���Ļ��ּ������4��3��2��Monkhorst-Pack(MP) k�����硣�ڷ����в��������µľ���������Ϊ�۹����Li (1s2 2s1)��O (2s2 2p4)��P (3s2 3p3)��Fe (3d6 4s2)��Ni (3d8 4s2)��Mg (2p6 3s2)�����Ƚ��м����Ż�����������������С�������µĹ��ͣ�������ƽ��͡������Ż���BFGS�㷨���������ǣ���������ı��������ֵdE/ion=1.0��10-5 eV��ԭ�Ӽ���������������ֵ|F|max=0.3 eV/nm��ԭ��λ�Ƶ�������ֵ|dR|max=1.0��10-4 nm��ԭ�Ӽ���Ӧ����������ֵ|S|max=5.0��10-2 GPa���Խṹ�Ż���Ľṹ�����ܴ�������̬�ܶȡ����ӵȷ�������̽��LiFePO4�ĵ绯ѧ�������۽ṹ�Ĺ�ϵ��
2 ������������
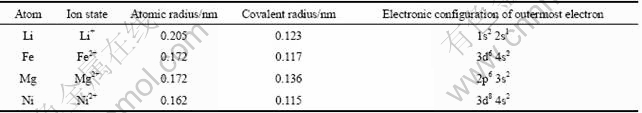
��1����Ϊ����LiFePO4�л���Ԫ�ؼ�����Ԫ�ص����ܣ����������ڹ��ɽ����������ڵ�Ԫ�أ�þ�����ڹ��ɽ�����������d������Ӳ��ڳɼ���ȡ����Mg[12-13]��Ni[24-26]Ϊ���ӽ���������ʱ��Ϊ+2�ۣ���һ�����������ֽ������Ӳ��ӶԵ��ӽṹ��Ӱ�죬��̽�ֵ绯ѧ���ܱ仯������ԭ�������Ӱ뾶���Ǻ�����ӵ�Ӱ�졣
���ݾ��飬Mg�Ĺ��۰뾶�ȱȱ�����Ԫ��Li��Fe�ĴȽ��Ѳ��ӣ�������Feλ����Ni�Ĺ��۰뾶�ȱ�����Ԫ��Li��Fe�Ķ�С��Ӧ�ñ�Mg���ײ��ӡ�
2.1 Ni����
ģ�����õ���ƫ̬�ܶ����ܴ�ͼ��ͼ2��3��ʾ�����Կ�����Ni���Ӻ����۲��ӷ�������Feλ����Liλ���ڷ����棬Ni-p�����Ni-s��������ú�С����ҪΪd������ӵ����á�
��һ���Բ���ǰ������渽�����ܴ�ͼ(��ͼ3)����������Feλ����Niʱ��Li���ٵ��µ��ܼ�(-44 eV����)���ܴ������٣���-22~-3 eV֮�䣬��LiFe7/8Ni1/8PO4�ĵ�9~136����Li7/8Ni1/8FePO4�ĵ�8~135������ΪP��O��s��p������ö��ɣ������������ϻ�����״�Ͼ��ϴ�仯�������������н��͡��ܴ��ı仯��Ҫ�ڷ����渽�������մ�϶ȡ�۴����͵����͵�ԭ����LiFe7/8Ni1/8PO4��ȡ��160�͵�163���ܴ������϶���õ�2.146 eV���ȴ�LiFePO4�Ĵ�϶(2.127 eV)�����������������ڼ۴����͵�����֮��Ľ����в����������µ����ʵ���(��161~162��)����һ���̶��������ڵ��ӵ絼������Li7/8Ni1/8Fe- PO4��ȡ��159�͵�165���ܴ������϶���õ�2.080 eV����϶���ͣ����ҽ�������5���м�� (��160~164��)����ˣ�Liλ���ӽ��������ڲ��ϵ��ӵ絼�ʵ����ӡ���Զ��ԣ�Ni������Liλ����Feλ��ʹ���ϵĵ���������߸��࣬ͬʱ��߳�ŵ����ʡ�
�о�����[13]������Ni2+���ӵ�LiFePO4�����ӷ�����Liλ������Ѳ�����(3%)�£���ŵ�����ܶ�Ϊ20 mA/gʱ��δ���ӺͲ���Ni��LiFePO4���״ηŵ�������ֱ�Ϊ116.34��145.80 mA��h/g��ѭ��20�κ���Ӧ�ķŵ�������ֱ�Ϊ102.26��138.66 mA��h/g����������������Ϊ��20�ηŵ���������״ηŵ������֮�ȣ���δ���ӺͲ���Ni��LiFePO4�����������ʷֱ�Ϊ87.90%��95.10%������ŵ�����ܶ����ӵ�320 mA/gʱ������Ni��LiFePO4��������������Ϊ84.41%��
2.2 Mg����
��Mg���ӵ�LiFePO4ƫ̬�ܶ�ͼ(��ͼ4)���Կ�����Mg���Ӻ����۲��ӷ�������Feλ����Liλ������Mg�ĺ���ɼ��������d������ӣ�Mg-s��������ú�С��Mg-p���Ҳ���Ե�������ã��Է����渽���ܴ������á�
��Mg����ǰ����ܴ�ͼ(��ͼ5)���������۲��ӷ�����Feλ����Liλ�����������µĵ��������λ����仯����Feλ����Mgʹ��϶���ӣ������ڵ��ӵ絼��Liλ����Mg�����ڴ�϶�Ľ��ͣ�ͬʱ�뵼��������p�ͱ��n�ͣ��ܼ�Ҳ��һ�����ͣ���ˣ�Liλ����Mg���������ڲ��ϵ��ӵ絼�ʵ����ӡ�
��1 ����LiFePO4�и�Ԫ�صĻ�������
Table 1 Basic properties of elements in doped LiFePO4
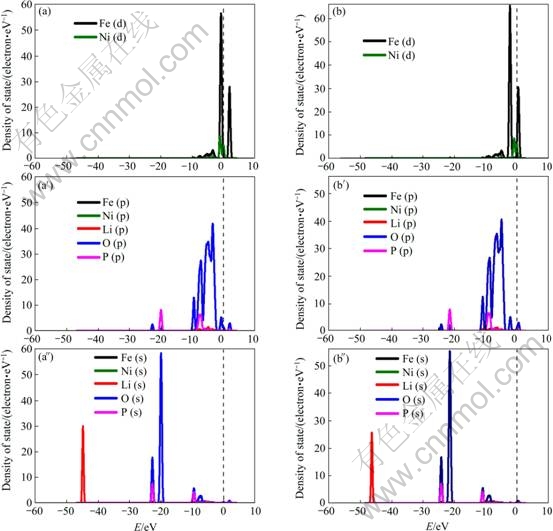
ͼ2 Ni����LiFePO4��ƫ̬�ܶ�ͼ
Fig. 2 Partial density of states of Ni-doped LiFePO4: (a), (a��), (a��) LiFe7/8Ni1/8PO4; (b), (b��), (b��) Li7/8Ni1/8FePO4
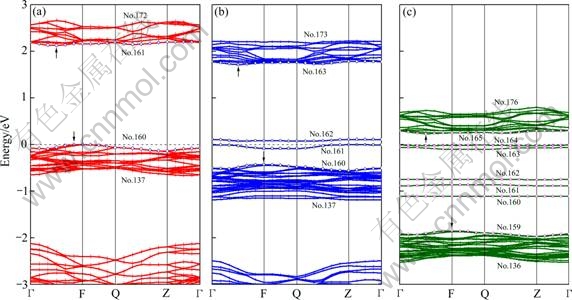
ͼ3 Ni����LiFePO4�ķ������ܴ�ͼ
Fig. 3 Band structure of Ni-doped LiFePO4 at Fermi level: (a) LiFePO4, band gap 2.127eV; (b) LiFe7/8Ni1/8PO4, band gap 2.146 eV; (c) Li7/8Ni1/8FePO4, band gap 2.080 eV
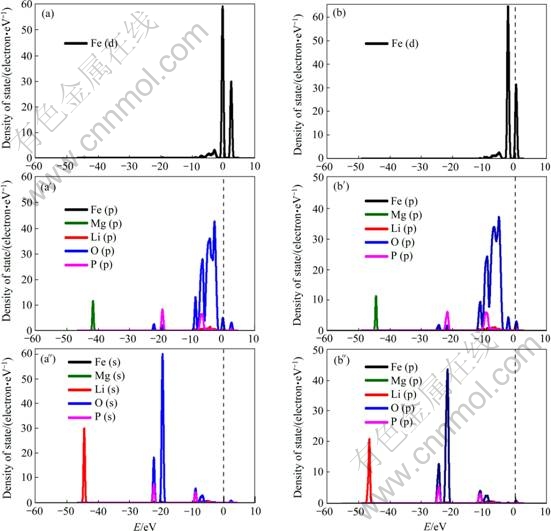
ͼ4 Mg����LiFePO4��ƫ̬�ܶ�ͼ
Fig. 4 Partial density of states of Mg-doped LiFePO4: (a), (a��), (a��) LiFe7/8Mg1/8PO4; (b), (b��), (b��) Li7/8Mg1/8FePO4
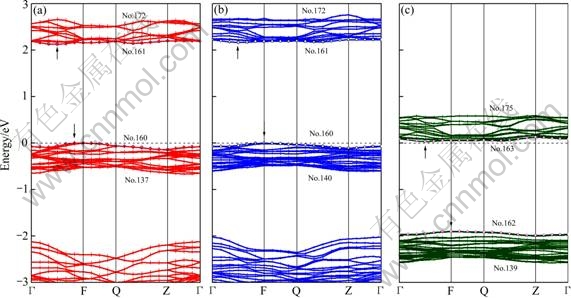
ͼ5 Mg����LiFePO4�ķ������ܴ�ͼ
Fig. 5 Band structure of Mg-doped LiFePO4 at Fermi level: (a) LiFePO4, band gap 2.127eV; (b) LiFe7/8Mg1/8PO4, band gap 2.166 eV; (c) Li7/8Mg1/8FePO4, band gap 1.932 eV
�����µ�[27]�о�����������Mg2+���ӵ�������ﮣ����ӷ�����Liλ������Ѳ�����(5%)�£���ŵ�����ܶ�Ϊ20 mA/gʱ���״ηŵ������Ϊ134.24 mA��h/g��ѭ��20�κ���Ӧ�ķŵ������Ϊ138.66 mA��h/g������������Ϊ91.63%������δ���ӵ�LiFePO4�ģ�����ŵ�����ܶ����ӵ�320 mA/gʱ������Mg��LiFePO4������������Ϊ66.20%��
��2��3����Ϊ��ͬλ�ò���Mg��Ni�ļ������Ƚϣ��������ܡ�������������϶�������ӷ���������ԭ�Ӽ����(rM��O)Ϊƽ��ֵ����ֵ���ܶ��ܱ�ʾ��ϵ�ڳɼ�ǰ����ԭ�ӹ����϶�����ĵ����ܶȷֲ��ı仯���Ӷ���Ӧ�ɼ������е��ӵ�ת�����������ԭ���������ֵ���ܶ�(![]() )��
)��
![]() (1)
(1)
ʽ�У�![]() Ϊ�ܵĵ����ܶȷֲ���
Ϊ�ܵĵ����ܶȷֲ���![]() Ϊ��ԭ�ӵ��Ǿ�������ܶȷֲ���
Ϊ��ԭ�ӵ��Ǿ�������ܶȷֲ���
ͼ6��ʾΪ��ͬλ�ò�ͬԪ�ز�������IJ�ֵ���ܶȱ仯�Ƚ�ͼ����ɫ��ʾ����ȱʧ����ɫ��ʾ���Ӹ���(�μ����Ӱ�)�����Կ������ڲ��������˲�ֵ���ܶȵı仯����ͬԪ�ء���ͬλ�ò��ӵ�Ӱ�������ͬ��Ni������Liλ��������ܶȵı仯�dz���Mg������Liλ����ı仯����Ni������Feλ��������ܶȵı仯����Ni��������ȱʧ��������Mg������Liλ����ĵ����ܶȱ仯���ԣ�ԭFe��O���Fe�����ǵ���ȱʧ����Mg����ȡ��Fe��Mg��O��Mg�����ǵ��Ӹ�����
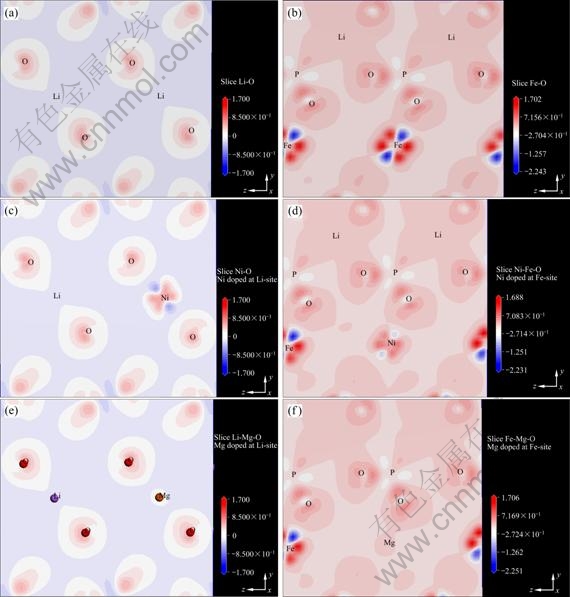
ͼ6 ��ͬλ�ò�ͬԪ�ز�������IJ�ֵ���ܶȱ仯ͼ
Fig. 6 Electron density difference maps at different positions with different elements: (a) Slice Li��O of undoped LiFePO4; (b) Slice Fe��O of undoped LiFePO4; (c) Slice Li��O of doped Li7/8Ni1/8FePO4; (d) Slice Fe��O of doped LiFe7/8Ni1/8PO4; (e) Slice Li��O of doped Li7/8Mg1/8FePO4; (f) Slice Fe��O of doped LiFe7/8Mg1/8PO4
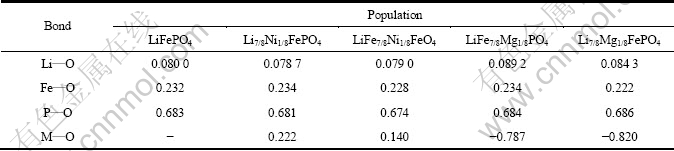
������ָ�����ڸ�ԭ�ӹ���ϵķֲ��������������������˽������ԭ�ӵijɼ������ͨ���ֱ�ԭ�Ӳ��Ӻͼ��������������ۡ������Ӱ���ͬһ������ܲ��ӣ���ͬ������ص����ӣ�ͨ�������ӷ������Եõ�ԭ�Ӽ����ľ������ݣ��Ӷ��ж��ɼ���ǿ�������ɹ��Ƽ��Ĺ����Ժ������Դ�С��һ��ߵIJ���ֵ�������ʹ����ԣ����͵IJ���ֵ������֮��Ϊ��������á���3�м�����(Population)����������Ni������Liλʱ��Ni��O����ֵΪ0.140������Li��O�IJ���ֵ(0.080)��Ni������Feλ������ֵΪ0.222��С�� Fe��O�IJ���ֵ(0.232)��Mg���۲�����Liλ����Feλ��Mg��O����ֵ��Ϊ��(-0.820��-0.787)��С�ڱ�ȡ��ԭ�ӵģ����������Լ�ǿ�������Լ�������������ӵ�صĹ���ԭ����Liλ�����Եļ�ǿ������Li��ɢ�����Mg������Liλ�����ڵ绯ѧ���ܵ���ߡ����ӷ����Ͳ�ֵ���ܶȱ仯��һ�¡�
һЩ�о�����Ϊ[21]�����ڲ������ӵİ뾶��Fe2+��С�������¾�����������ʵ�ϣ������Ӿ����У��������Ӽ�ĺ˼��Ϊ�������Ӱ뾶֮�ͣ����������ӵĽ����ںδ������ж������ⲻͬ���͵ľ����У�ͬ�����ӱ��ֳ����İ뾶��һ�������������ۣ�Li+��1s���ȫ����2s���ȫ�գ��γ�s�ӻ������Fe2+��3d�������4���۵��ӣ�Ni2+��3d�������2���۵��ӣ�Mg2+��3s���ȫ�գ�2p���ȫ�����γ�sp3�ӻ�������ڱ�ģ���У����Ƽ���ѡȡ�ļ۵������ԭ�ӵ���������Ų���һ�¡���2�в��Ӻ�ľ���������Ȳ���ǰ���ӣ���Ҫ�����ھ��������ĸı䡣��Ȼ������ӣ����ӱ�����������ֵ���Կ��������Ӻ�ϵͳ�����������ڲ���ǰ��˵�����Ӻ����ϵ���ȶ�������ʹ���ϵij�ŵ�ѭ�����ܸ��á����Ӻ���ϵ�ľ������������
��2��ԭ�Ӽ�ƽ������(r)��ʵ�����ݾ�Ϊ����������Խ��������ģ��IJ���������ʵ����������Լ�ʵ�鼰���Ե����ص�Ӱ�죬�������һ���IJ� ��Fe��O���ۼ���ǿ������Fe��O��P���յ�Ч Ӧ���Լ�Fe��P֮��ľ����ų����ö�������ͬʱҲʹFe3+/Fe2+������ԭ��Զ���Li�ķ����ܼ���·ѭ����ѹ(OCV)�ܸߣ�OCV��Ҫ�ܾ���������PO43-��Ӱ��[28]������������PO4ʹLiFePO4�ṹ�ȶ�����������Fe3+/Fe2+������ԭ��Եķ����ܼ����Ӷ����ӵ缫��λ�������IJ��Ӳ���Ӱ��OCV��Feλ���� ʱ��P��O������С(��0.000 1 nm)��Liλ����ʱ��P��O���Ҳ������0.000 1��0.000 2 nm��˵���ṹ�ܽ��ա�
ʵ������������������Ӻ�Li��Oԭ�Ӽ��ƽ�������δ���ӵ����ӡ���ˣ����������Ľ������Ӻ�Li��O��������ӣ�ʹ�����֮��Ľ����������������������ӵ���Ƕ(��ɢ�˶�)���Ӷ�����������﮾��бȽϺõĴ�����ŵ����ܡ����⣬������˵���������Ӳ�����Liλ��ʱ�����ڽ������Ӳ�����ɢ�����ܻ��Li���ӵ���ɢ��һ�����谭���á��ɱ�2��֪��Ni������Liλʱ��M��O֮�����ֱ�Ϊ0.217 3��0.218 3 nm����Li��O֮�����Ϊ0.212 8��0.211 5 nm����rM��O��rLi��O��������Li���ӵ���ɢ�˶�����Mg������Liλʱ��������෴��M��O��Li��O֮�����ֱ�Ϊ0.210 1��0.211 6 nm����rLi��O��rM��O�������Ni���Ӹ�������Li+����ɢ�˶�����ˣ������ӷ�����Liλʱ�������ϸ���Ʋ�������
��2 ��ͬ����Ԫ�صļ������Ƚ�
Table 2 Comparison of calculation results of different doped LiFePO4
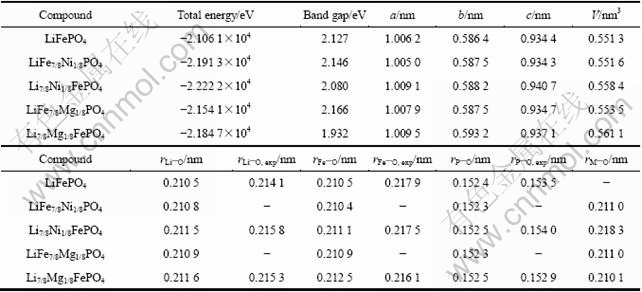
��3 ��ͬ����Ԫ�ؼ��IJ���ֵ
Table 3 Bond population of different doped element bands
���ϵij�ŵ������ܵ��Ӵ��ݵ�Ӱ�죬ȱ�ݵIJ���ʹ���ϵĵ��ӽṹ��������϶���뵼�����͡����ʴ��IJ����������Եȷ�����仯���Ӷ�Ӱ���ŵ����ܡ�������Ϸ������Եõ����½��ۣ�Ni�ĵ��ӽṹ����������������d����е�������˶�Ӱ������������ڸ��Թ�������������Ӱ��Զ����Mg�ȷǹ��ɽ���Ԫ�صģ�Ni�����Dz�����Liλ����Feλ�������ڵ��ӵ絼�ʵ�����ŵ����ʵ����ӡ�Mg������Liλʱ������϶�����ܽ��ͣ�Ҳ���������Li���ӵ���ɢ�˶�����Mg������Feλʱ�����ǵ��ӵ絼����Li������ɢ��û����ߣ��Բ��ϵ������ܺͳ�ŵ����ʵ�û�и��ơ���ˣ�ֻҪ��ʹ���ӵ絼�����ӣ���ʹ����ӵ���ɢ�˶����ӣ�������������Ϊ﮵���������ϵ�ʹ�á�ģ�������ʵ���о������������������Ľ������ӿ�����Ч�����������﮵ı�������ѭ�����ܺʹ�����ŵ����ܣ��ҹ��ɽ������Ӳ��Ӿ��нϺõĴ�����ŵ����ܡ�
3 ����
1) Ni������ĵ��ӽṹ����������������d����е�������˶�Ӱ���������������ڲ��Ӹ���ʱ���ṹ�ȶ�����϶����ʹ���ӵ絼�����ӣ���ŵ�������ߡ�
2) Mg���Ӹ���ʱ����������Liλ����϶�����ܾ����ͣ�ͬʱLi+����ɢ�˶�Ҳ��ߣ������ڸ��ԣ�����Feλ����ʱ�������渽��Fe-d����е���Ӱ���ܴ���ʹ��϶���ӣ������ڵ��ӵ絼�����IJ��ӷ���Ҳ����Li��O��������ǿ��������������ɢ���������ڸ��ԡ�
3) ���ۺ��ֲ���ȱ�ݣ�ֻҪ��ʹLiFePO4��d����ĵ��ӷ����仯���Ӷ��Բ��ϵ��۽ṹ����Ӱ�죬����Ӱ����绯ѧ���ܡ������۽ṹ��Ҫ�Ƿ����渽�����ܴ����ر�����d����е�������˶���Ӱ�졣
���
���ϴ�ѧ��ѧ�뻯��ѧԺΪ�������ṩ����֧�֡�������ѧ�½�������Ϊ���������ݷ����ṩ�dz���Ҫ�����ۺͽ��顣�������õ����Ϻ������������ĵ�֧�֡�
REFERENCES
[1] PADHI A K, NANJUNDASWAMY K S, GOODENOUGH J B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries[J]. J Electrochem Soc, 1997, 144(4): 1188-1194.
[2] CHUNG S Y, BLOCKING J T, CHIANG Y M. Electronically conductive phospho-olivines as lithium storage electrodes[J]. Nat Mater, 2002, 1: 123-128.
[3] NAKAMURA T, SAKUMOTO K, OKAMOTO M, SEKI S, KOBAYASHI Y, TAKEUCHI T, TABUCHI M, YAMADA Y. Electrochemical study on Mn2+-substitution in LiFePO4 olivine compound[J]. J Power Sources, 2007, 174(2): 435-441.
[4] NAKAMURA T, MIWA Y, TABUCHI M, YAMADA Y. Structural and surface modifications of LiFePO4 olivine particles and their electrochemical properties[J]. J Electrochem Soc, 2006, 153(6): A1108-A1114.
[5] ZHOU F, KANG K, MAXISCH T, CEDER G, MORGAN D. The electronic structure and band gap of LiFePO4 and LiMnPO4[J]. Solid State Commun, 2004, 132(3/4): 181-186.
[6] ZHOU F, COCOCCIONI M, MARIANETTI C A, MORGAN D, CEDER G. First-principles prediction of redox potentials in transition-metal compounds with LDA+U[J]. Phys Rev B: Condens Matter, 2004, 70(23): 235121.
[7] MORGAN D, VAN DER VEN A, CEDER G. Li conductivity in LixMPO4 (M=Mn, Fe, Co, Ni) olivine materials[J]. Electrochem Solid-State Lett, 2004, 7(2): A30-A32.
[8] WANG C S, HONG J. Ionic/electronic conducting characteristics of LiFePO4 cathode materials[J]. Electrochem Solid-State Lett, 2007, 10(3): A65-A69.
[9] �߽���, �ܺ��, �¼���, �չ�ҫ. �����Ӳ��Ӷ�LiFePO4��Ӱ��[J]. ������ѧѧ��, 2004, 20(6): 582-586.
NI Jiang-feng, ZHOU Heng-hui, CHEN Ji-tao, SU Guang-yao. Effect on the electrochemical performance of lithium iron phosphate by Cr3+ ion doping[J]. Acta Phys-Chim Sin, 2004, 20(6): 582-586.
[10] �߽���, �ܺ��, �¼���, ������. ������������Ӹ���LiFePO4�绯ѧ����[J]. ����ѧѧ��, 2005, 21(4): 472-476.
NI Jiang-feng, ZHOU Heng-hui, CHEN Ji-tao, ZHANG Xin-xiang. Improvement of LiFePO4 electrochenmical performance by doping metal oxides[J]. Chinese Journal of Inorganic Chemistry, 2005, 21 (4): 472-476.
[11] �� ��, ������, �ڴ���, �Ķ���, ������. ����Mo��LiFePO4�������ϵĵ绯ѧ����[J]. ������ѧѧ��, 2008, 24(8): 1498-1502.
CHEN Yu, WANG Zhong-li, YU Chun-yang, XIA Ding-guo, WU Zi-yu. Electrochemical properties of Mo-doped LiFePO4 cathode material[J]. Acta Phys-Chim Sin, 2008, 24(8): 1498-1502.
[12] TENG T H, YANG M R, WU S H, CHIANG Y P. Electrochemical properties of LiFe0.9Mg0.1PO4/carbon cathode materials prepared by ultrasonic spray pyrolysis[J]. Solid State Commun, 2007, 142(7): 389-392.
[13] ������, ������, ������, ������, ������, ��ǭ��, ���ٿ�. ��ѧ�������Ʊ�����������﮵Ľṹ�������о�[J]. ϡ�н��������빤��, 2007, 36(6): 954-958.
ZHANG Pei-xin, WEN Yan-xuan, LIU Jian-hong, XU Qi-ming, REN Xiang-zhong, ZHANG Qian-ling, LUO Zhong-kuan. Structure and performance of doped lithium iron phosphate by chemical precipitation[J]. Rare Metal Materials and Engineering, 2007, 36(6): 954-958.
[14] CEDER G, CHIANG Y M, SADOWAY D R, AYDINOL M K, JANG Y I, HUANG B. Identification of cathode materials for lithium batteries guided by first-principles calculations[J]. Nature, 1998, 392: 694-696.
[15] MAXISCH T, CEDER G. Elastic properties of olivine LixFePO4 from first principles[J]. Phys Rev B: Condens Matter, 2006, 73: 174112.
[16] WANG L, ZHOU F, MENG Y S, CEDER G. First-principles study of surface properties of LiFePO4: Surface energy, structure, Wulff shape, and surface redox potential[J]. Phys Rev B: Condens Matter, 2007, 76: 165435.
[17] �˻���, ������, ����, ����Ӱ, ��·�. LiNiO2�����ӻ�����Ľṹ���ȶ��Եĵ�һ��ԭ���о�[J]. ���ӿ�ѧѧ��, 2007, 23(2): 99-103.
GU Hui-min, WANG Dong-lai, ZHAI Yu-chun, LIU Li-ying, LI De-fa. The first-principle study on the structures and stabilities of LiNiO2 and its doping compounds[J]. Journal of Molecular Science, 2007, 23(2): 99-103.
[18] ����Ӱ, �����, �˻���, ����. ���õ�һԭ�������о�Al3+����LiMn2O4�ĵ��ӽṹ[J]. ���ӿ�ѧѧ��, 2008, 24(3): 207-209.
LIU Li-ying, SHEN Hong-tao, GU Hui-min, ZHAI Yu-chun. The first principle investigation of the electronic structure of Al3+ doped LiMn2O4[J]. Journal of Molecular Science, 2008, 24(3): 207-209.
[19] OUYANG C Y, SHI S Q, WANG Z X, HUANG X J, CHEN L Q. First-principles study of Li ion diffusion in LiFePO4[J]. Phys Rev B: Condens Matter, 2004, 69: 104303.
[20] OUYANG C Y, SHI S Q, WANG Z X, LI H, HUANG X J, CHEN L Q. The effect of Cr doping on Li ion diffusion in LiFePO4 from first principles investigations and Monte Carlo simulations[J]. J Phys: Condens Matter, 2004, 16: 2265-2272.
[21] WANG Z L, SUN S R, XIA D G, CHU W S, ZHANG S, WU Z Y. Investigation of electronic conductivity and occupancy sites of Mo doped into LiFePO4 by ab initio calculation and X-ray absorption spectroscopy[J]. J Phys Chem C, 2008, 112(44): 17450-17455.
[22] MOMMA K, IZUMI F. VESTA: A three-dimensional visualization system for electronic and structural analysis[J]. J Appl Crystallogr, 2008, 41(3): 653-658.
[23] ANDERSSON A S, KALSKA B, H GGSTR M L, THOMAS J O. Lithium extraction/insertion in LiFePO4: An X-ray diffraction and M?ssbauer spectroscopy study[J]. Solid State Ionics, 2000, 130(1/2): 41-52.
[24] PARAGUASSU W, FREIRE P T C, LEMOS V, LALA S M, MONTORO L A, ROSOLEN J M. Phonon calculation on olivine-like LiMPO4 (M=Ni, Co, Fe) and Raman scattering of the iron-containing compound[J]. J Raman Spectrosc, 2005, 36(3): 213-220.
[25] LU Y, SHI J, GUO Z, TONG Q, HUANG W, LI B. Synthesis of LiFe1-xNixPO4/C composites and their electrochemical performance[J]. J Power Sources, 2009, 194(2): 786-793.
[26] FISHER C A J, PRIETO V M H, ISLAM M S. Lithium battery materials LiMPO4 (M=Mn, Fe, Co, and Ni): Insights into defect association, transport mechanisms, and doping behavior[J]. Chem Mater, 2008, 20: 5907-5915.
[27] ������, ������, ������, ������, ������, ��ǭ��. þ���Ӳ��Ӷ�������﮽ṹ�����ܵ�Ӱ��[J]. ���ܲ���, 2006, 37(12): 1942-1945.
ZHANG Pei-xin, WEN Yan-xuan, LIU Jian-hong, XU Qi-ming, REN Xiang-zhong, ZHANG Qian-ling. Performance and structure of Mg2+ doped lithium iron phosphate prepared by chemical precipitation method[J]. J Func Mater, 2006, 37(12): 1942-1945.
[28] WANG G X, BEWLAY S, NEEDHAM S A, LIU H K, LIU R S, DROZD V A, LEE J F, CHEND J M. Synthesis and characterization of LiFePO4 and LiTi0.01Fe0.99PO4 cathode materials[J]. J Electrochem Soc, 2006, 153(1): A25-A31.
(�༭ ������)
������Ŀ��������Ȼ��ѧ����������Ŀ(50874074��50474092)���㶫ʡ��Ȼ��ѧ����������Ŀ(8151806001000028)�������пƼ��ƻ�������Ŀ (ZYC200903250150A��200505)�������й��ܸ߷����ص�ʵ���ҿ��Ż���������Ŀ(FP20110004)
�ո����ڣ�2011-08-21�������ڣ�2011-11-20
ͨ�����ߣ������£����ڣ���ʿ���绰��0755-26558134��E-mail: pxzhang2000@163.com
ժ Ҫ�����û����ܶȷ������۵ĵ�һ��ԭ���о�����ӵ����������LiFePO4����Ni��Mg�ĵ��ӽṹ�����������Ni������ĵ��ӽṹ������������d����е���Ӱ�죬���ڲ��Ӹ���ʱ���ṹ�ȶ�����϶����ʹ���ӵ絼�����ӣ���ŵ�������ߣ�Mg���Ӹ���ʱ����������Liλʱ����϶�����ܾ����ͣ�ͬʱLi���ӵ���ɢ�˶�Ҳ��ߣ������ڸ��ԣ�����Feλ����ʱ�������渽��Fe-d����е���Ӱ���ܴ���ʹ��϶���ӣ������ڵ��ӵ絼�����IJ��ӷ���Ҳ����Li��O��������ǿ��������������ɢ���������ڸ��ԡ���ˣ�ͨ�����ӿɶ�LiFePO4���۽ṹ����Ӱ�죬�Ӷ�Ӱ����绯ѧ���ܣ��������۽ṹ��Ҫ��d����е����˶��Է����渽���ܴ���Ӱ�죬����������ӵĻ��ϼۺͰ뾶�ء�
