
���±�ţ�1004-0609(2008)01-0118-08
Sb��Bi�Ͻ����Mg-Alϵ�Ͻ�������ܵĻ���
�ܵ���1������ˮ2��¬Զ־1���ų���1
(1. ���ϴ�ѧ ���������Ƚ������������ص�ʵ���ң���ɳ 410082��
2. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410082)
ժ Ҫ��
���û����ܶȷ������۵�Dmol 4.1���ӺϽ��γ��ȡ�����ܡ�����ѧ���ܺ�̬�ܶȵȷ��棬�о�Sb��Bi�Ͻ����Mg-Alϵ�Ͻ�������ܵ�Ӱ����������������Sb��Bi�ֱ��û�Mg-Alϵ�Ͻ�Mg17Al12����Mg(��)��Mg(��)��Mg(��)��Alԭ��ʱ����Sb�û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�����γ��ȶ���Mg17Al12������ṹ�������Sb��Bi��Mg17Al12�й��������ޡ���һ���ȽϺϽ��γ��ȶ��Ĺ�����ṹ������Sb��Bi�Ͻ�����ṹ���ȶ��Ա�δ�Ͻ�ʱ��ǿ�����У�Sb�û�Mg17Al12����Mg(��)ԭ��ʱ����ṹ�ȶ�����ߣ����Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ�Bi�û�Mg17Al12����Mg(��)ԭ�ӣ������������仯����Mg3Bi2��Mg3Sb2������Ӧ�Ͻ�Mg17Al12������Ľṹ���ȶ�����ͬ�¶�������ѧ���ܵļ��㷢�֣��Ͻ���ϵ���γ��˽ṹ�ȶ��Ըߵ�Sb��Bi�Ͻ�Mg17Al12�������Լ������仯����Mg3Sb2��Mg3Bi2����Щ��ߵĽṹ�ȶ��Բ������¶ȵ����߶���ʧ����ṹ�ȶ����Ա�Mg17Al12��ߣ����Sb��Bi�Ͻ������Mg-Alϵ�Ͻ�Ŀ�������ܡ�����̬�ܶȵķ��������һ��������Mg-Alϵ�Ͻ�����ṹ�ȶ�����ߵ���Ҫԭ�����ڣ�Sb��Bi�Ͻ���ϵ�����ܼ����µ��ܼ����ɼ������������࣬����Դ��Ҫ��Mg(s)��Mg(p)��Al(p)��Bi(d)��Sb(d)�ļ۵��ӡ�
�ؼ��ʣ�
Mg17Al12�����ṹ�ȶ���������ѧ������
��ͼ����ţ�TG 146.2���� ���ױ�ʶ�룺A
Mechanism of Sb, Bi alloying on improving heat resistance properties of Mg-Al alloy
ZHOU Dian-Wu1, LIU Jin-shui2, LU Yuan-zhi1, ZHANG Chu-hui1
(1. State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body, Hunan University,
Changsha 410082, China;
2. School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: By using Dmol 4.1 program based on the density functional theory, heat of formation, cohesive energy, thermodynamic properties and density of states (DOS) of the alloying system were investigated to explain the mechanism of the influence of Sb, Bi alloying on improving heat resistance properties of Mg-Al alloy. The results show that the structure of these phases can exist and be stable when the Mg atoms at the positions ��, �� of the Mg17Al12 phase are substituted with Sb, the Mg atoms at the positions �� are substituted with Bi, which shows that Sb and Bi are little soluted in Mg17Al12 phase. By comparing the stable Mg17Al12 phase with Sb or Bi addition, it is found that Mg17Al12 phase stability is improved, and Mg17Al12 solid solutions substituting Mg atoms at positions �� with Sb has the highest structural stability, next substituting Mg atoms at positions �� with Sb, finally substituting Mg atoms at positions �� with Bi, whereas the structures of Mg3Sb2 and Mg3Bi2 are more stable than that of Mg17Al12 solid solutions with Sb, Biadditions. By calculating the thermodynamic properties of Mg-Al alloy, it is found that the improved heat resistance properties of the alloying system are caused by forming the Mg17Al12 phase and intermetallic Mg3Sb2 and Mg3Bi2 with higher structural stability, which is not changed with the elevated temperature. Compared with the density of states (DOS) of the alloying system, the increase of the structural stability of Mg-Al alloy with Sb or Bi additions attributes to an increase in the bonding electron numbers at lower energy level below Fermi level, which mainly originates from the contribution of the valence electron numbers of Mg(s), Mg(p), Al(p), Bi(d) and Sb(d) orbits.
Key words: Mg17Al12 phases; structural stability; thermodynamic properties
�������ѳ�Ϊ������ҵ��չ�ı�Ȼ���ƣ���ΪĿǰ����Ĺ��̽�������֮һ������Ϊ��21������ɫ���̲��ϡ���þ�Ͻ���������ҵ�ϵ�Ӧ�õõ��˳���ķ�չ��Ȼ��Ŀǰ������þ�Ͻ�ֻ���ڳ����·��۵Ŀ����֧��������������������������ʹ��������еõ�Ӧ�ã���˿��������¶ȴ���150 ��Ŀ��������þ�Ͻ��Ϊ����þ�Ͻ��о�������ȵ㡣
Ӧ���������ϵ�þ�Ͻ��㲿�������ѹ��������Mg-Alϵ�Ͻ�ռѹ��þ�Ͻ�����90%���ϡ��úϽ�ϵ��Ҫ��a-Mg��������b-Mg17Al12���������[1]������b-Mg17Al12���ڸ�������������������Ч������������Ƹ��¾���ת�������¸���ǿ�ȡ�������� ���½�, ���������Mg-Alϵ�Ͻ��Ӧ�á�Ŀǰ�� �����ڸ����¾����������ܵ�þ�Ͻ����Dz���RE��Ag��Y��Th�ȹ�����Ͻõ��ģ�ֻ�������������ɻ��������ĸ����ܷ�������Ӧ�ã���û�������������ϵõ��ƹ�Ӧ�á�Ԭ������[2?3]��Mg-Alϵ�Ͻ��м����������۽���Ԫ��Sb��Bi������Bi���γɽ����仯����Mg3Bi2�⣬��ƫ���ھ��磬�ٽ�������������Mg17(Al, Bi)12������������˺Ͻ���ϵ�����ȶ��ԣ���Sb������Mg17Al12�����Mg3Sb2��ʽ����������Mg-Al�Ͻ���¿�������ܵ�������������о������Mg-Alϵ�Ͻ���ۺ����������Ǹ��¿�������ܷ���ȡ���˱Ƚ������ �����
���ڽ�ʾ�Ͻ����Mg-Al�Ͻ���¿�������ܵ�Ӱ�����ʮ�ָ��ӣ�Ŀǰ���������ױ���Ҳ�� ��[4?6]������չ�ⷽ��������о���ָ����������������ʮ����Ҫ����ʵ���塣Ϊ�ˣ��������߲��û����ܶȷ������۵�Dmol 4.1�������ѡȡ�п�����ҵ����Sb��Bi�Ͻ�Mg-Al�Ͻ�ϵ����Sb��Bi�Ͻ�ǰ����ϵ�ĺϽ��γ��ȡ�����ܡ�����ѧ���ܺ�̬�ܶȵȷ��棬����ȷ��Sb��Bi������Mg17Al12���γ��ȶ��Ĺ�����ṹ��Ȼ���һ��̽�ֹ�����ṹ�Լ�Mg-Al�Ͻ�ϵ���������仯����Mg3Sb2��Mg3Bi2�ڲ�ͬ�¶�����ṹ���ȶ��ԣ����ӵ��Ӳ�εĽǶȣ���ͼ��ʾSb��Bi�Ͻ����Mg-Alϵ�Ͻ�������ܵ�Ӱ�������
1 ����ģ���뷽��
1.1 ����ģ��
����Mg-Alϵ�Ͻ�������ܲ����Ҫԭ��������ϵ��Mg17Al12���ڸ��������������ṹ�ȶ��ԲΪ�˱��ļ���ѡ��A12��Mg17Al12����Ϊ�о������侧��ṹ��ͼ1(a)��ʾ��������a=b=c=1.057 97 nm���ռ�ȺΪI![]() 3m����߶Գ���ΪTd3��������ԭ������Ϊ58����ԭ������Ϊ
3m����߶Գ���ΪTd3��������ԭ������Ϊ58����ԭ������Ϊ
+2Mg(��): (0, 0, 0), (1/2, 1/2, 1/2)
+8Mg(��): (x, x, x), (?x, ?x, x) x=0.32
+24Mg(��): (x, x, z), (?x, ?x, z), (?x, x, ?z), (x, ?x, ?z) x=0.36 z=0.04
+24Al: (x, x, z), (?x, ?x, z), (?x, x, ?z), (x, ?x, ?z) x=0.09 z=0.28
�������ԭ��ģ�ͣ�ԭ���ṹ��ͼ1(b)��ʾ��ԭ������Ϊ29������12��Alԭ�Ӻ�17��Mgԭ��(�ֱ�Ϊ1��Mg(��)��4��Mg(��)��12��Mg(��)ԭ��)��Sb��Bi�Ͻ�ʱ�����ǵ�Ԭ������[2?3]ʵ�鷢��Sb��Bi������Mg17Al12�����γɹ�����ṹ�����˷ֱ���1��Sb��Bi�û�Mg17Al12ԭ���е�1��Mg(��)��4��Sb��Bi�û�4��Mg(��)��12��Sb��Bi�û� 12��Mg(��)��12��Sb��Bi�û�12��Al����Ӧ�� ��(Mg16Sb)Al12��(Mg16Bi)Al12��(Mg13Sb4)Al12��(Mg13Bi4)Al12��(Mg5Sb12)Al12��(Mg5Bi12)Al12��Mg17Sb12��Mg17Bi12ԭ��ģ�͡�
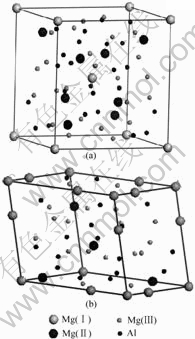
ͼ1 Mg17Al12��ľ�����ԭ���ṹ
Fig.1 Mode of cell(a) and primitive cell(b) of Mg17Al12 phase
1.2 ���㷽��
���IJ��û����ܶȷ������۵�Dmol 4.1������������Ż������ܼ���ʱ�����ӽ��������ܺ�������LDA���Ƶ�PWC��ʽ[7]���ƺ���ȡȫ����λ�ƣ����Ӳ���������DND������[8]������Ԩ�����ֲ���Monkhorst-Pack��ʽ������K�㷽��[9]����������ǰ�Ƚ��м����Ż�����ȡ��ģ�͵ľ����ȶ��ṹ���Ż�ʱ�侫������Ϊ��������1.0��10?5 Ha��Ӧ����0.004 Ha��λ�ơ�0.000 5 nm������Ͻ���ϵ��ͬ�¶��µ�����ѧ���ܣ����ó����е�Dynamicsģ�顣ѡȡNVT�����ӽ��������ܺ�������GGA���Ƶ�BLYP��ʽ[5]���ƺ���ȡȫ����λ�ƣ����Ӳ��������ô�d�����˫��ֵ��(DNP) ����������Fine����ɢ���Smearing energy������������������
2 ���������
2.1 �Ͻ��γ���
���IJ�����ʽ����Sb��Bi�Ͻ�ǰ��Mg17Al12ԭ��ģ��ƽ��ÿ��ԭ�ӵĺϽ��γ���(?H) [10?11]��

��̬��ԭ����������ʱ�����������ԭ����������ͬ���ƺ�����Mg��Al��Sb��Bi���嵥ԭ�������ļ���ֵ�ֱ�Ϊ��?199.125 5��?241.465 4��?6 310.531 5 ��?20 090.531 5 Ha������õ�Sb��Bi�Ͻ�ǰ��Mg17Al12ԭ��ģ�͵ĺϽ��γ�����ͼ2(a)��ʾ����ͼ2(a)�ɼ���Mg17Al12��ĺϽ��γ���Ϊ?0.052 eV/atom��������[12]�����Ľ��?0.034 eV/atom�ӽ�������Sb�û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�ӣ��Ͻ���ϵ�ĺϽ��γ���Ϊ��������Sb��Bi�Ͻ�Sb�û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�����γ��ȶ���Mg17Al12������ṹ[13]����Sb�û�Mg17Al12����Mg(��)��Alԭ�ӣ�Bi�û�Mg17Al12����Mg(��)��Mg(��)��Alԭ��ʱ���Ͻ���ϵ�ĺϽ��γ���ȴΪ�����������γɵ�Mg17Al12������ṹ���ȶ���Sb��Bi��Mg17Al12�еĹ��������ޣ�����������ʱ�����γɵڶ�������仯���
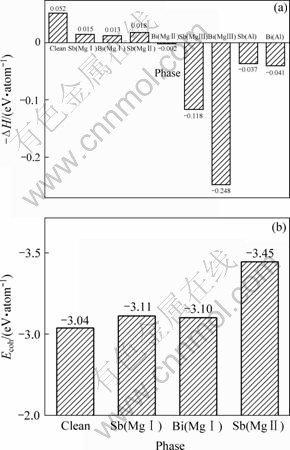
ͼ2 Sb��Bi�Ͻ�ǰ��Mg17Al12��ĸ��Ͻ��γ���(?��H)�ͽ����(Ecoh)
Fig.2 Negative formation heat (?��H)(a) and cohesive energy (Ecoh)(b) of Mg17Al12 phase with and without Sb or Bi addition (Clean, Sb(Mg��), Bi(Mg��), Sb(Mg��), Bi(Mg��), Sb(Mg��), Bi(Mg��), Sb(Al), Bi(Al) denote Mg17Al12, (Mg16Sb)Al12, (Mg16Bi)Al12, (Mg13Sb4)Al12, (Mg13Bi4)Al12, (Mg5Sb12)Al12, (Mg5Bi12)Al12, Mg17Sb12 and Mg17Bi12 phases, respectively)
Ԭ������[2?3]��Mg-Alϵ�Ͻ����Sb��Bi�Ͻ����ֳ�������������Mg17(Al,Bi)12�����⣬Bi���γ��˽����仯����Mg3Bi2����Sb��������Mg17Al12�⣬���γ��˽����仯����Mg3Sb2��Ԭ������ʵ����[2?3]֤ʵ�������������ó��Ľ����Ϊ�ˣ�����ʽ(2)�����˽����仯����Mg3Sb2��Mg3Bi2�ĺϽ��γ���(?��H)��
����Mg3Sb2�ĺϽ��γ���Ϊ?0.418 5 eV/atom(Լ?40.38 kJ/mol)����ʵ��ֵ[14] ?16.0 kcal/mol(Լ?66.944 kJ/mol)�ȽϽӽ�����Mg3Bi2�ĺϽ��γ���Ϊ?0.326 eV/atom(Լ?31.453 7 kJ/mol)��������˵����Ԭ������[2?3]����Sb��Bi������Mg17Al12�࣬���γ��ȶ��Ĺ�����ṹ����Bi�γ�Mg17(Al,Bi)12������ṹȴ���ȶ������Ͻ���ϵ�����Ľ����仯����Mg3Sb2��Mg3Bi2����ṹ�ȶ����ڡ�
2.2 �����
Ϊ��һ���Ƚ�Sb, Bi�Ͻ�Mg-Alϵ�Ͻ��γ������ṹ�ȶ�������Ľṹ�ȶ��Դ�С�����ķֱ����ʽ(3)��(4)��������Sb�ֱ��û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�����γɽṹ�ȶ����ڵ�Mg17Al12������Ľ����(Ecoh)�Լ������仯����Mg3Bi2��Mg3Sb2�Ľ����(![]() )[11]��
)[11]��

Mg��Al��Sb��Bi����ԭ�������ļ���ֵ�ֱ�Ϊ��?199.125 5��?241.298��?6 310.349 9��?20 090.369 2 Ha������õ�Mg17Al12��Sb��Bi�Ͻ�ǰ��Ľ������ͼ2(b)��ʾ����ͼ2(b)�ɼ���Sb��Bi�Ͻ�ǰ���ֱ��û���ϵ�е�Mg(��)��Mg(��)ԭ�Ӻ�Bi�û�Mg17Al12����Mg(��)ԭ�ӣ�Sb�û�Mg17Al12����Mg(��)��Mg(��)ԭ�ӣ�ԭ����ϵ�Ľ���������������ھ���Ľ��ǿ��ͨ���ý���ܱ�ʾ[15]��������ܾ��ǽ�����ԭ�ӽ��Ϊ�������ͷŵ�������Ҳ���ǰѾ���ֽ�ɵ���ԭ������Ҫ���Ĺ��������Խ���γɾ���Խ�ȶ�[15]������Mg17Al12��������ṹ�࣬�Ͻ��γ�(Mg16Bi)Al12��(Mg16Sb)Al12��(Mg13Sb4)Al12����ֽ������ԭ������Ҫ������Խ�࣬����Sb��Bi�Ͻ�Mg17Al12��ϵ�Ľṹ�ȶ�����ǿ[16?17]�����У�Sb�û�Mg(��)ԭ��ʱ����ṹ���ȶ������Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ���Bi�û�Mg17Al12����Mg(��)ԭ�ӣ�����(4)ʽ����õ������仯����Mg3Bi2��Mg3Sb2�Ľ���ֱܷ�Ϊ��?3.579 4 eV/atom��?3.375 3 eV/atom��������ṹ����Ӧ�Ͻ�Mg17Al12������Ľṹ���ȶ���
2.3 ����ѧ����
2.2�ڵļ�����ֻ������̬(��0 K) ʱ��Sb��Bi�Ͻ������Mg17Al12��Ľṹ�ȶ��ԣ���Mg3Bi2��Mg3Sb2�Ľṹ������Ӧ�Ͻ�Mg17Al12������Ľṹ���ȶ�����Ԭ��������Mg-Alϵ�Ͻ��м���Ԫ��Sb��Bi��ʵ���¶Ȳ���Ϊ0 K��������Щ��Ľṹ�ȶ����ڸ���ʱ����Mg17Al12��ǿ�أ���ṹ�ȶ��Ի�����¶ȵ����߶���ʧ�أ�Ϊ��һ����ʾSb��Bi�Ͻ����Mg-Alϵ�Ͻ���¿�������ܵ�Ӱ����������ļ����˺Ͻ���ϵ��ͬ�¶���(��298~573 K)������ѧ���ܣ�Sb��Bi�Ͻ�Mg17Al12��������ϵ����(S)����(H)�ļ������ֱ���ͼ3(a)��ͼ3(b)��ʾ��
��ͼ3(a)��ͼ3(b)�ɼ�����������(298 K)���ߵ�300 ��(573 K)ʱ��Sb��Bi�Ͻ�Mg17Al12��ϵ����(S)������(H)���١�����Gibbs������Խ�ͣ���ϵ��ṹ���ȶ������ã�Ϊ�ˣ���һ������ʽ(5)������Sb��Bi�Ͻ�Mg17Al12��������ϵ��Gibbs������(G)��
![]()
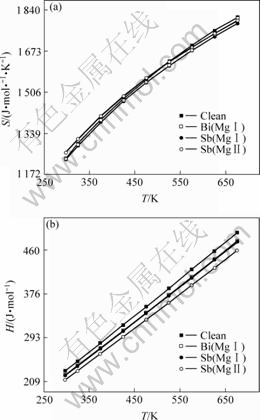
ͼ3 Sb��Bi�Ͻ�ǰ��Mg17Al12�ͬ�¶��µ�������
Fig.3 Entropy(S)(a) and Enthalpy(H)(b) of Mg17Al12 phase under different temperatures with and without Sb or Bi addition (Clean, Sb(Mg��), Bi(Mg��), Sb(Mg��) denote Mg17Al12, (Mg16Sb)Al12, (Mg16Bi)Al12, (Mg13Sb4)Al12 phases, respectively)
��������ͼ4(a)��ʾ����ͼ�ɼ��������¶ȵ����ߣ�Sb��Bi�Ͻ�Mg17Al12��ϵ��Gibbs�����ܾ���δ�Ͻ�ʱ���٣�����Sb�û�Mg(��)ԭ��ʱ��Mg17Al12��ϵ�ṹ��Gibbs��������С�����Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ���Bi�û�Mg17Al12����Mg(��)ԭ�ӡ��������Sb��Bi�Ͻ�Mg17Al12��������ϵ�Ľṹ�ȶ��Բ������¶ȵ����߶������仯�����Ͻ���ϵ�Ľṹ�ȶ��Ծ���δ�Ͻ�ʱǿ�����У�Sb�û�Mg(��)ԭ��ʱ�ṹ���ȶ�����ߣ����Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ���Bi�û�Mg17Al12����Mg(��)ԭ�ӡ�����Sb��Bi�Ͻ������Mg17Al12���ڸ����µĽṹ���ȶ��ԣ���Щ��Ľṹ���ȶ��Բ������¶ȵ����߶���ʧ����Mg17Al12�������ڸ����½ṹ���ȶ��Ե���ߣ���������������˸���������Ч������������Ƹ��¾���ת���������Mg-Alϵ�Ͻ�Ŀ�������ܡ�
Mg-Al�Ͻ��У�Ԭ������[2?3]���ּ���Ԫ��Sb��Biʱ�������˽����仯����Mg3Bi2��Mg3Sb2��2.1�ڼ��������������ֽ����仯����ṹ�ȶ����ڣ�2.2�ڼ�����������ṹ����Ӧ�Ͻ�Mg17Al12������Ľṹ���ȶ�������Mg-Alϵ�Ͻ����¶�һ��Ϊ150~300 �棬Ϊ�˱��Ľ�һ������ͱȽ���Mg17Al12�������仯����Mg3Sb2��Mg3Bi2��ͬ�¶��µ�Gibbs�����ܡ���ͼ4(b)��ʾ��Mg3Sb2��Mg3Bi2��Gibbs�����ܾ���Mg17Al12��ĵͣ�����Mg3Sb2��Mg3Bi2�Ľṹ���ȶ����ڸ����¾���Mg17Al12��ߣ�����Щ��Ľṹ�ȶ���Ҳ�����¶ȵ����߶��仯��
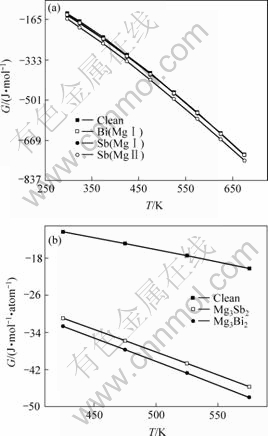
ͼ4 Sb��Bi�Ͻ�ǰ��Mg17Al12�ͬ�¶��µ�Gibbs�����ܺ�Mg17Al12��Mg3Bi2��Mg3Sb2��ͬ�¶���ƽ��ÿ��ԭ�ӵ�Gibbs������
Fig.4 Gibbs free energy of Mg17Al12 phase under different temperatures with and without Sb or Bi addition and Per atom Gibbs free energy of Mg17Al12 phase, Mg3Bi2 and Mg3Sb2 under different temperatures (Clean, Sb(Mg��), Bi(Mg��)��Sb(Mg��) denote Mg17Al12, (Mg16Sb)Al12, (Mg16Bi)Al12, (Mg13Sb4)Al12 phases, respectively)
���ڽṹ���ȶ��Ը��ߵ�Mg3Sb2��Mg3Bi2�Ĵ��ڣ����Mg-Al�Ͻ���ϵ����ǿ�ȡ����������ߡ�������������ȻSb��Bi�Ͻ����Mg-Al�Ͻ���¿�������ܵ�Ӱ�����ʮ�ָ��ӣ������ĵļ�����һ���̶��ϣ��Ϻý�����Ԭ������[2?3]��Mg-Alϵ�Ͻ��м���Ԫ��Sb��Bi���Ͻ���ϵ�����������ߵ�ʵ������
2.4 ̬�ܶ�
��2.3������ѧ���ܵļ�������֪��Sb��Bi�Ͻ����Mg-Alϵ�Ͻ�������ܵ���Ҫԭ�����ڣ���ϵ���γ��˲����¶����߶���ʧ���ṹ���ȶ��Ըߵ�Sb��Bi�Ͻ�Mg17Al12�������Լ������仯����Mg3Sb2��Mg3Bi2��Ϊ��һ������Sb��Bi�Ͻ����Mg-Alϵ�Ͻ���ṹ�ȶ��ĵ��ӻ��ƣ�����ƪ�����ƣ����Ľ�ѡȡBi��Sb�Ͻ��γɽṹ�ȶ���Mg17Al12��������Ϊ�о���������Mg17Al12��(Mg16Bi)Al12��(Mg16Sb)Al12��(Mg13Sb4)Al12�������̬�ܶ���ֲ�̬�ܶȣ���ͼ5��ʾ����ͼ�ɼ����Ͻ�ǰ(ͼ5(a))����0.1~?0.35 Ha��Χ�ڣ�Mg17Al12��ijɼ�������Ҫ��Mg(s)��Mg(p)��Al(s)�� Al(p)�ļ۵��ӹ��ף�Bi�Ͻ�(ͼ5(b))����0.1~?0.35 Ha��Χ�ڣ�(Mg16Bi)Al12�ijɼ����ӳ�����Mg(s)��Mg(p)��Al(s)�ļ۵��ӹ����⣬Bi(p)�ļ۵�����һ���Ĺ��ף�����?0.35~?0.40 Ha�ķ�Χ�ڣ�������һ������Bi(d)�۵��ӹ����³ɼ��壻Sb�Ͻ��û�MgIԭ��(ͼ5(c))����0.1~?0.35 Ha��Χ�ڣ�(Mg16Sb)Al12�ijɼ�������Ҫ����Mg(s)��Mg(p) ��Al(p) �ļ۵��ӹ��ף�����?0.35~?0.40 Ha��Χ���γɵ��³ɼ��壬ȴ����Mg(s)��Mg(p)�ļ۵��ӹ��ף�Sb�Ͻ��û�MgIIԭ��(ͼ5(d))����0.1~?0.35 Ha��Χ�ڣ�(Mg13Sb4)Al12�ijɼ����ӳ�����Mg(s)��Mg(p) ��Al(p) �ļ۵��ӹ����⣬Sb(s)��Sb(p)��Sb(d)�ļ۵������൱��Ĺ��ף�������?0.35~?0.40 Ha��Χ�ڣ��³ɼ���ĸ߶�������������Al(p)��Sb(d)�Ĺ��ס�
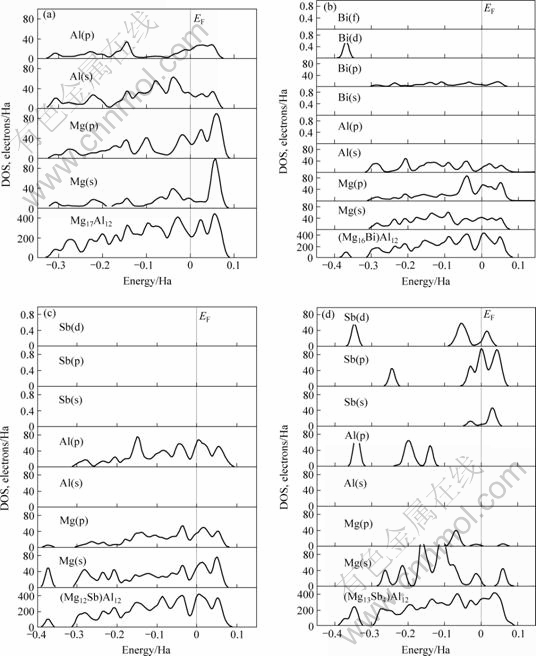
ͼ5 Sb��Bi�Ͻ�ǰ��Mg17Al12���̬�ܶ�
Fig.5 DOS of Mg17Al12 phase with and without Sb or Bi addition: (a) Mg17Al12; (b) (Mg16Bi)Al12; (c) (Mg16Sb)Al12; (d) (Mg13Sb4)Al12
��һ���Ƚ�ͼ6�ɼ���Mg17Al12�����Ҫ�ɼ���ֲ���0.1~?0.35 Ha��Χ�ڣ�Bi�Ͻ��û�Mg��ԭ�Ӻ�(Mg16Bi)Al12��Ҫ�ɼ�����Ȼ�ֲ���0.1~?0.35 Ha��Χ�ڣ��ҳɼ���ĸ߶ȱ仯������?0.35~?0.40 Ha��Χ�ڣ�������һ���³ɼ��壬��ֵ�߶�Ϊ��107.142 9������״̬/Ha����Mg17Al12��Bi�Ͻ��ڵ��ܼ����ɼ����������࣬���Bi�Ͻ���ϵ�ṹ��ø����ȶ�[18?19]����Sb�Ͻ��û� Mg��ԭ�Ӻ�(Mg16Sb)Al12����Ҫ�ɼ���ķֲ���Χ��ɼ���߶���Bi�Ͻ��������ƣ���˺Ͻ���ϵ�ṹͬ����ø����ȶ�����Sb�Ͻ��û�Mg��ԭ�ӣ�(Mg13Sb4)Al12����Ҫ�ɼ�����Ȼ��Ȼ��0.1~?0.35 Ha��Χ�ڣ����ɼ���ĸ߶������ر�����?0.35~?0.40 Ha�ķ�Χ�ڣ�������һ�����ߵ��³ɼ��壬��ֵ�߶ȴ�243.622 4������״̬/Ha����δ�Ͻ�Bi��Sb��
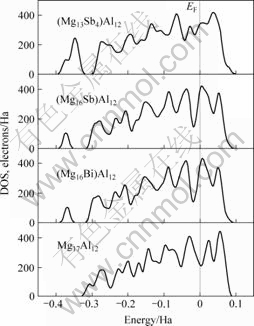
ͼ6 Sb��Bi�Ͻ�ǰ��Mg17Al12���̬�ܶ�
Fig.6 DOS of Mg17Al12 phase with and without Sb or Bi addition
���û�MgIԭ�Ӻ����̬�ܶ���ȣ���0.1~?0.35 Ha��?0.35~?0.40 Ha�ķ�Χ�ڣ�(Mg13Sb4)Al12��ɼ��������������ࡣ����һ����ɼ����������������۵����������ǿ����һ�������ijɼ�����λ�ڵ��ܼ�����ʹ��ϵ�ṹ��ø����ȶ������Sb�Ͻ��û�Mg��ԭ�Ӻ���ϵ�ṹ�ͳ������ȶ��Ľṹ[18?19]��
3 ����
1) ���������ܶȷ�������Dmol 4.1���������Mg-Al�Ͻ�ϵSb��Bi�Ͻ�ǰ����ϵ�ĺϽ��γ��ȣ�����ܡ�����ѧ������̬�ܶȡ�
2) Sb��Bi�ֱ��û�Mg17Al12����Mg(��)��Mg(��)��Mg(��)��Alԭ��ʱ����Sb�û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�����γ��ȶ���Mg17Al12������ṹ��Sb��Bi��Mg17Al12�еĹ��������ޡ�
3) Sb��Bi�Ͻ��γ�Mg17Al12�����壬��ṹ����δ�Ͻ�ʱ�ȶ�������Sb�û�Mg17Al12����Mg(��)ԭ��ʱ����ṹ�ȶ�����ߣ����Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ���Bi�û�Mg17Al12����Mg(��)ԭ�ӣ����������仯����Mg3Bi2��Mg3Sb2�Ľṹ������ӦSb��Bi�Ͻ�Mg17Al12������Ľṹ���ȶ���
4) Sb��Bi�Ͻ����Mg-Alϵ�Ͻ���¿�������ܵ���Ҫԭ�����ڣ��Ͻ���ϵ���γ��˲����¶����߶���ʧ���ṹ�ȶ��Ը���Mg17Al12��ĺϽ�Mg17Al12�������Լ������仯����Mg3Sb2��Mg3Bi2��
5) Mg-Alϵ�Ͻ�����Ľṹ�ȶ�����ߵ���Ҫԭ����Sb��Bi�Ͻ���ϵ�����ܼ����µ��ܼ��� �ɼ������������࣬����Դ����Ҫ��Mg(s)��Mg(p)��Al(p)��Bi(d)��Sb(d)�۵��ӡ�
[1] LUO A, PEKGULERYUZ M O. Cast magnesium alloys for elevated temperature application[J]. J Mater Sci, 1994(20), 29: 5259?5271.
[2] Ԭ����, ��С��, ������, ���Ľ�, ������. Sb�Ͻ�þ�Ͻ���ѧ���ܵĸ�������[J]. ���Ϲ���, 2001(4): 10?15.
YUAN Guang-yin, ZENG Xiao-qing, L? Yi-zhen, DING Wen-jiang, SUN Yang-shan. Mechanical properties improvement of Mg-Al based alloy with Sb addition[J]. Mater Eng, 2001(4): 10?15.
[3] Ԭ����, ��Ϊ��, ������. Mg-Al���Ͻ��Bi�Ͻ�����ѧ���ܵĸ�������[J]. ���ϴ�ѧѧ��, 1999, 29(3): 115?119.
YUAN Guang-yin, ZHANG Wei-min, SUN Yang-shan. Effect of Bismuth addition on the mechanical properties of Mg-Al based alloys[J]. Journal of Southeast University, 1999, 29(3): 115?119.
[4] �Ź�Ӣ, �� ��, ������, ���Ų�. Bi,Sb�Ͻ�AZ91þ�Ͻ���֯������Ӱ������о�[J]. ����ѧ��, 2005, 54(11): 5288?5292.
ZHANG Guo-ying, ZHNAG Hui, FANG Ge-liang, LI Yu-cai. A study on the mechanism of the influence of Bi, Sb alloying on microstructure and properties of AZ91 magnesium alloy[J]. Acta Physica Sinica, 2005, 54(11):5288-5292
[5] DU Wen-wen, SUN Yang-shan, MIN Xue-gang, et al. Influence of Ca addition on valence electron structure of Mg17Al12[J]. Chinese Nonferrous Metals, 2003, 13(6): 1247?1280.
[6] ��ѧ��, ������, Ѧ ��, ��. Ca���Mg17Al12���۵������EET���۷���[J]. ��ѧͨ��, 2002, 47(2): 109?112.
MIN Xue-gang, DU Wen-wen, XUE Feng, et al. The melting point of Mg17Al12 phase improved by alloyed with Ca and analysis based on the empirical electron theory (EET)[J]. Chinese Sci Bull, 2002, 47 (2): 109?112.
[7] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18/28): 3865?3868.
[8] PACK J D, MONKHORST H J. Special points for Brillouin- zone integrations��A reply[J]. Phys Rev B, 1977, 16(4/15): 1748?1749.
[9] DELLEY B. Analytic energy derivatives in the numerical local-density-functional approach[J]. J Chem Phys, 1991, 94(11): 7245?7250.
[10] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, et al. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl[J]. Acta Mater, 1998, 46(10): 3433?3442.
[11] SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Mater Sci Eng B, 1997, 49(1): 74?78.
[12] ZHOU D W, PENG P, LIU J S. Energetics, electronic structure and structure stability of the calcium alloying Mg17Al12 phase from first principles calculations[J]. Mater Sci, 2007, 25(1): 145?153.
[13] SONG Y, GUO Z X, YANG R, et al. First principles study of site substitution of ternary elements in NiAl[J]. Acta Mater, 2001, 49(9): 1647?1654.
[14] LI C, HOE J L, WU P. Empirical correlation between melting temperature and cohesive energy of binary laves phases[J]. J Phys Chem Solids, 2003, 64(2): 201?212.
[15] ZUBOV V I, TRETIAKOV N P, TEIXEIRA RABELO J N, SANCHEZ ORTIZ J F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a van der Waals crystal-fullerene C60[J]. Phys Lett A, 1995, 198(5/6): 470.
[16] ISHII Y, FUJIWARA T. Electronic structures and cohesion mechanism of Cd-based quasicrystals[J]. Non-cryst Solids, 2002, 312?314(12): 494?497.
[17] FAGAN S B, MOTA R, BAIERLE R J, et al. Stability investigation and thermal behavior of a hypothetical silicon nanotube[J]. J Molecular Struct, 2001, 539(1): 101?106.
[18] FU C L, WANG X D, YE Y Y. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallics, 1999,7(2): 179?184.
[19] NYLEN J, GARCIA F J, MOSEL B D. Structure relationships, phase stability and bonding of compounds PdSnn (n=2, 3, 4)[J]. Solid State Sci, 2004, 6(1): 147?155.
������Ŀ����������ʿ�����������Ŀ(20020530012)�����ϴ�ѧ�����˲Ż���������Ŀ(20068339828)
�ո����ڣ�2007-05-08�������ڣ�2007-09-13
ͨѶ���ߣ��ܵ��䣬�����ڣ���ʿ���绰��13017297124��E-mail: ZDWe_mail@yahoo.com.cn
ժ Ҫ�����û����ܶȷ������۵�Dmol 4.1���ӺϽ��γ��ȡ�����ܡ�����ѧ���ܺ�̬�ܶȵȷ��棬�о�Sb��Bi�Ͻ����Mg-Alϵ�Ͻ�������ܵ�Ӱ����������������Sb��Bi�ֱ��û�Mg-Alϵ�Ͻ�Mg17Al12����Mg(��)��Mg(��)��Mg(��)��Alԭ��ʱ����Sb�û�Mg17Al12����Mg(��)��Mg(��) ԭ�ӣ�Bi�û�Mg17Al12����Mg(��)ԭ�����γ��ȶ���Mg17Al12������ṹ�������Sb��Bi��Mg17Al12�й��������ޡ���һ���ȽϺϽ��γ��ȶ��Ĺ�����ṹ������Sb��Bi�Ͻ�����ṹ���ȶ��Ա�δ�Ͻ�ʱ��ǿ�����У�Sb�û�Mg17Al12����Mg(��)ԭ��ʱ����ṹ�ȶ�����ߣ����Sb�û�Mg17Al12����Mg(��)ԭ�ӣ��ٴ�Bi�û�Mg17Al12����Mg(��)ԭ�ӣ������������仯����Mg3Bi2��Mg3Sb2������Ӧ�Ͻ�Mg17Al12������Ľṹ���ȶ�����ͬ�¶�������ѧ���ܵļ��㷢�֣��Ͻ���ϵ���γ��˽ṹ�ȶ��Ըߵ�Sb��Bi�Ͻ�Mg17Al12�������Լ������仯����Mg3Sb2��Mg3Bi2����Щ��ߵĽṹ�ȶ��Բ������¶ȵ����߶���ʧ����ṹ�ȶ����Ա�Mg17Al12��ߣ����Sb��Bi�Ͻ������Mg-Alϵ�Ͻ�Ŀ�������ܡ�����̬�ܶȵķ��������һ��������Mg-Alϵ�Ͻ�����ṹ�ȶ�����ߵ���Ҫԭ�����ڣ�Sb��Bi�Ͻ���ϵ�����ܼ����µ��ܼ����ɼ������������࣬����Դ��Ҫ��Mg(s)��Mg(p)��Al(p)��Bi(d)��Sb(d)�ļ۵��ӡ�
