
DOI: 10.11817/j.ysxb.1004.0609.2021-37829
ʯīϩ����Lin(n<10)������Ϊ�ĵ�һ��ԭ������
�� ��1,2��������2������ΰ1����˼��1���� ��1���� ΰ#1�����ܱ�#1
��1 �������̼�����ѧ ���Ͽ�ѧ�빤��ѧԺ������ ���� 123000��
2 �������̼�����ѧ ��ѧ�빤��ѧԺ������ ���� 123000)
ժ Ҫ��
���û����ܶȷ����ĵ�һ��ԭ���������о�������ӵ��ʯīϩ����Liԭ�ӻ��ž���Lin(n<10)�ij�����Ϊ�������������Liԭ����n��2ʱ��Liԭ�������Է�ɢ����ʽ������ʯīϩ̼��Ԫ�������Ϸ�����n��3ʱ��Liԭ���������ž������ʽ������ʯīϩ���棬�����γ��ȶ���Li4��Li7��Li9�ž��塣�����ʯīϩ�������������ܳ���ʯī���������γ��֦����Liԭ�ӵĵ���ת�����ٽ�ʯīϩ�ķ����й����Liԭ�ӵ�2s�����Cԭ�ӵ�2p����������Ե��ӻ���������n������ϵ�ķ����ܼ���ʯīϩ�����й���ƶ��������Ժ͵��ӵ�������ǿ��Lin�ž���ײ��Li-Li��ͨ��Ϊ���Ӽ���������Li-Li��ͨ��Ϊ���ۼ���Li-C��Ϊ���в��ֹ��ۼ����Ե����Ӽ�������Li-C��ǿ������n����������С��
�ؼ��ʣ�
����ӵ����ʯīϩ��Lin�ž�������һ��ԭ����������Ϊ��
��ͼ����ţ�TM 912.9 ���ױ�־�룺A ���±�ţ�
����ӵ�ؾ��������ܶȴ�����ѹ��ѭ�����������ŵ㣬�ڱ�Яʽ�ƶ������豸�͵綯��������ҵ�õ��㷺Ӧ�á�����ӵ�صĴ��������缫�����йأ�ʯī�������Ͼ���ѭ���������ͳɱ��͵��ŵ㣬��Ŀǰ�г�����ߺ������о���ע���ĵ缫����֮һ�������۴������Ϊ372 mAh/g��������������ӵ�ضԸ�����������������������ӵ�ؼ���缫���ϳ�ΪĿǰ���ܼ��������о����ȵ�[1]��
ʯīϩ���г���ıȱ���������õĵ����Ժͷdz��ߵ���ѧǿ�ȣ��ڴ��ܵ缫�����о������õ�Ӧ��ǰ������ΪĿǰ����ӵ�ظ��������������о����ȵ�[2,3]�����侲��[4]�ϳɵĶ��ʯīϩ����30 mA/g�����ܶ��µ�������822 mAh/g�����Ҿ��нϸߵ�ѭ�����ܺͱ������ܡ���ʫ����[5]����AFM�������������о�ʯīϩ��Ƕ���Ϊ�������������Ҫ��ʯīϩ�����ɢ�����ڲ����ɢ���衣
��ӱ��[6]���õ�һ��ԭ�������о���Li��ʯīϩ�����������Ǩ����Ϊ������Li��ɢ·����Ҫ�ǿ�ԽC-C���ţ���ɢ����Ϊ0.336 eV��Zhang��[7]�о���ʯīϩ��������ӵ���ɢϵ�������ݣ��Ƶ�������Ԥ�ⷽ�̡�Yildirim��[8]�����о���ʯīϩȱ�ݶ�Liԭ�ӵ�Ӱ�죬����Stone-Walesȱ�ݺ�˫��λȱ�ݶ�Li�Ľ��ǿ�ȸ��ߣ���������ȱ���ܶȻ��һ����ǿ��Li������á�Liu��[9]�����о����֣�Bȡ������ʯīϩ������Ч��ߴ����������״C3B����������Ϊ714 mAh/g��ԼΪʯī���������������Rani��[10]���㷢�֣�B��N���ӿ��Ե���Li��ʯīϩ֮��ļ������Ժͽ���ܡ�Koh��[11]�����о���Li��ʯīϩ-����ϩ�ӻ���ϵ�ϵ����������������ӻ���ϵ��Li��������ʯīϩ�ȶ�������Li���������ڸ���ϩ�ࡣDavid��[12]���㷢�֣�ʯīϩ��C3N4�γɵ����ʽ��ܹ���ɢ���������Liԭ�ӣ����Ҿ��п��ٵĵ��ת�����������⣬ʯīϩҲ������������ӵ�ص����͵����[13,14]��
�����о���Ҫ����������ʯīϩ����ȱ�ݡ�Ԫ�ز����Լ����ϵ�;�������ʯīϩ�Ĵ��������Liԭ����Ҫ�Է�ɢ��ʽ�ij�����ʯīϩ���棬��ʯīϩ���������Liԭ������ֱ����أ���Ŀǰȱ��ʯīϩ������ž��������Ϊ������о������IJ��û����ܶȷ����ĵ�һ��ԭ���������о�������ӵ��ʯīϩ��������Liԭ�ӻ��ž���Lin(n<10)������������͡������ܡ�����ܶȵ�ɺ�̬�ܶȵ����ܣ�����Ϊ�������ӵ��ʯīϩ�������ϵ�Ƕ������ṩ����ָ����
1 ���ۼ��㷽��
������ӵ��ʯīϩ�����ij������У�Liԭ��������ʯīϩ���棬��Ӧ��������ΪG+nLi Lin-G������GΪ6��6��ʯīϩ������nΪLiԭ��������Lin-GΪʯīϩ�������Liԭ�ӻ�Lin(n<10)�ž����γɵĻ�������û����ܶȷ������۵�ƽ�沨���Ʒ�����CASTEP������[15]��Lin-G�����������������͡������ܡ�����ܶȵ�ɺ�̬�ܶȵ����ʽ������ۼ��㡣��������н��������ܲ��ù����ݶȽ���(GGA)�µ�PBE�����ͷ��»�������Grimme����[16]��ͨ�������Բ��ԣ�ƽ�沨չ���Ľض�����Ϊ500 eV��Monkhorst-Pack��k������ֲ�[17]��Ϊ5��5��1��ԭ�ӵ���������������Ϊ1.0��10-5 eV/atom������������Ϊ0.2 eV/nm��Ϊ�����������Ծ���֮���Ӱ�죬��ղ�ѡȡ2 nm������õ�6��6��ʯīϩ������C-C����Ϊ0.1420 nm���������һ��[18]��
Lin-G������GΪ6��6��ʯīϩ������nΪLiԭ��������Lin-GΪʯīϩ�������Liԭ�ӻ�Lin(n<10)�ž����γɵĻ�������û����ܶȷ������۵�ƽ�沨���Ʒ�����CASTEP������[15]��Lin-G�����������������͡������ܡ�����ܶȵ�ɺ�̬�ܶȵ����ʽ������ۼ��㡣��������н��������ܲ��ù����ݶȽ���(GGA)�µ�PBE�����ͷ��»�������Grimme����[16]��ͨ�������Բ��ԣ�ƽ�沨չ���Ľض�����Ϊ500 eV��Monkhorst-Pack��k������ֲ�[17]��Ϊ5��5��1��ԭ�ӵ���������������Ϊ1.0��10-5 eV/atom������������Ϊ0.2 eV/nm��Ϊ�����������Ծ���֮���Ӱ�죬��ղ�ѡȡ2 nm������õ�6��6��ʯīϩ������C-C����Ϊ0.1420 nm���������һ��[18]��
Ϊ���о�ʯīϩ�������Liԭ�ӻ�Lin�ž����γɻ����������ѧ�ȶ��ԣ�����������[19]Ϊ��
��Eab=[E(Lin-G)-E(G)-nE(Li)]/n (1)
����E(G)��E(Li)��E(Lin-G)�ֱ�Ϊʯīϩ������������������Liԭ�ӵ�������ʯīϩ�������Liԭ�ӻ�Lin�ž����Lin-G�������������������EabΪ��ֵ������Liԭ�ӻ�Lin�ž����ܹ��ȶ�������ʯīϩ���档
2 ���������
2.1 �������ͷ���
ʯīϩ�����������Liԭ�ӣ������������λ�ã��ֱ�Ϊ̼��Ԫ���������Ϸ�(��ΪHλ)��̼-̼���������Ϸ�(��ΪBλ)��̼ԭ�����Ϸ�(��ΪTλ)����ͼ1��ʾ��ͨ���ṹ�Ż����֣�Liԭ����Hλ������������Eab��ͣ�Ϊ-1.9030 eV����Bλ��Tλ�ġ�Eab�������ֱ�Ϊ-1.5282��-1.5275 eV������������������Liԭ�����ȳ�����ʯīϩHλ�������߶�h��ͣ�Ϊ0.1803 nm��Liԭ����ʯīϩ��������ã�����ʯīϩ̼ƽ�淢������λ�ƣ���hֵҲ��ͣ���Ϊ0.0009 nm����ʱ��ӦLi��Cԭ��֮���ƽ������ΪdLi-CΪ0.2295 nm���ӽ�ʯīǶ﮻�����LiC6��Li-C����(0.2331 nm)��
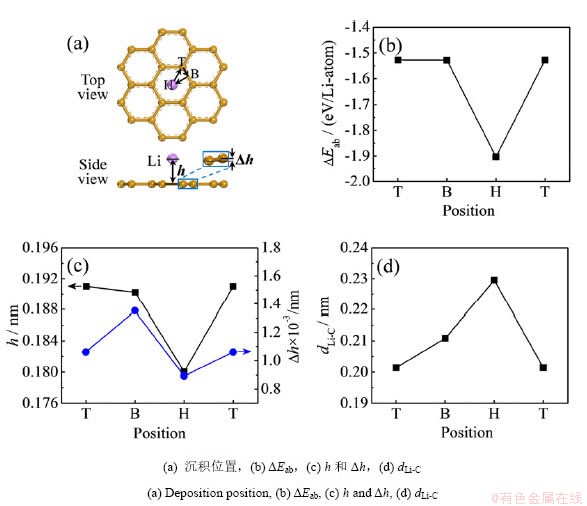
ͼ1 ʯīϩ���浥��Liԭ�ӵij�����Ϊ
Fig.1 Deposition behavior of single Li atom on graphene
��ʯīϩ�������2��Liԭ�ӣ����������Լ���Eab��h����h��dLi-C��Liԭ��֮�����Ĺ�ϵ��ͼ2������Liԭ��֮����������Eabֵ���Ͳ�����ƽ����hֵ�Ƚ��ͺ�С��������2��Liԭ�������Է�ɢ����ʽ������ʯīϩ̼��Ԫ�������Ϸ�λ�á���2��Liԭ�����һ��̼��Ԫ��������Ϊ0.5054 nmʱ��hֵ��С��Ϊ0.1776 nm����hΪ0.0014 nm����ʱ��ӦdLi-CֵҲ��С��Ϊ0.2282 nm���ȵ���Liԭ�ӳ�����ʯīϩHλ��dLi-C��С������2��Liԭ�������ֹ��ͳ�����ʯīϩ���棬Liԭ����ʯīϩ֮�������ý�ǿ��
ͼ2 ʯīϩ����2��Liԭ�ӵij�����Ϊ
Fig.2 The deposition behavior of 2 Li atoms on graphened
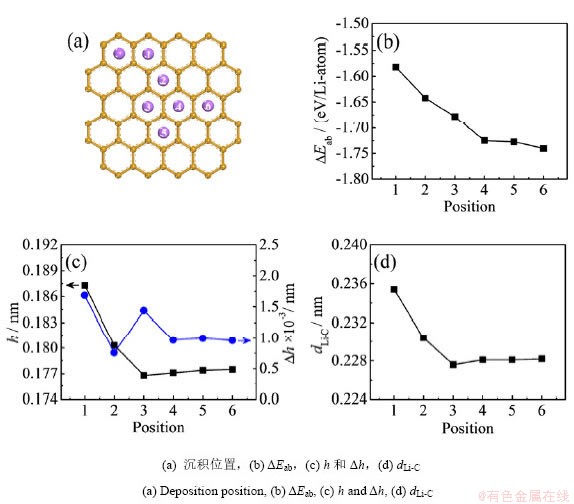
��ʯīϩ�������3��Liԭ�ӣ��������Ͱ����ž��幹�ͺͷ�ɢ���ͣ���ͼ3�������ܼ��������������ɢ������ȣ�3��Liԭ�������γ��ž��壬��ƽ����ʯīϩƽ��������ι��ͳ�����ʯīϩ���棬�����ܡ�Eab��ͣ�Ϊ-1.6769 eV����ͼ3(a)��ʾ��������������У�Liԭ�ӵ�ƽ�������߶�hΪ0.1938 nm���ȵ���Liԭ�Ӻ�2��Liԭ�ӵij���ƽ���߶ȸߣ�dLi-CֵΪ0.2414����LiC6��Li-C����(0.2331 nm)������dLi-LiֵΪ0.2771 nm���Ƚ���Li��Li-Li�����̣������ƽ����ʯīϩƽ��������ι���Li3�ž�����ʯīϩ֮�������ã��ȵ���Liԭ�ӻ�2��Liԭ�ӳ���ʱ�����������������
ͼ3 ʯīϩ����3��Liԭ�ӵij�������
Fig.3 The deposition configurations of 3 Li atoms on graphene
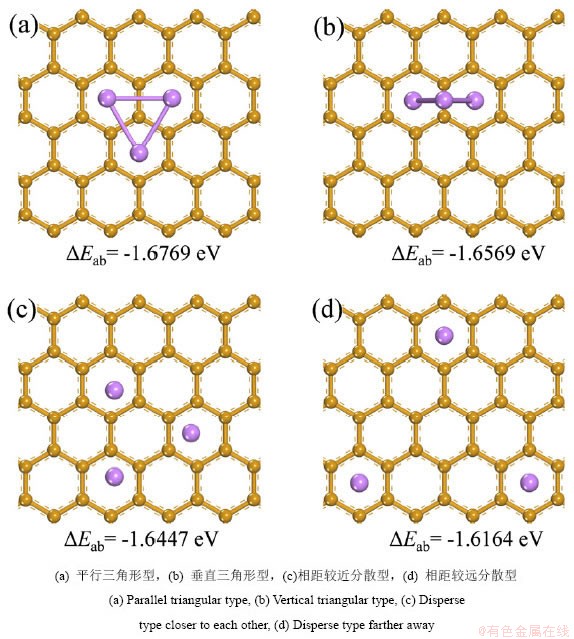
����ʯīϩ�������Liԭ�����������࣬��n��3ʱ��Liԭ�Ӿ����ž������ʽ������ʯīϩ���档��ͬLin�ž���������͵������ܡ�Eab���Լ�����������͵�h����h��dLi-C��dLi-Li��Liԭ������n֮��Ĺ�ϵ��ͼ4������Liԭ������n�����࣬Lin�ž��������������������ȴ�������Ӻ�����ƽ�ȵ����ƣ���n=4��7��9ʱ�������������Խϵͣ����������ӵ��ʯīϩ������ʵ�����п��ܳ����γ��ȶ���Li4��Li7��Li9�ž��塣
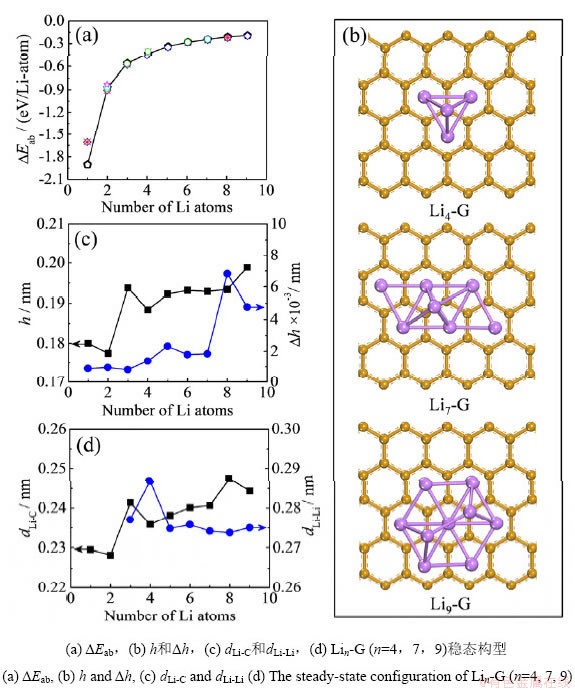
ͼ4 ʯīϩ����Lin�ž���ij�����Ϊ��Liԭ����֮��Ĺ�ϵ
Fig.4 The relationship between the deposition behavior of Lin aggregate on graphene and the number of Li atoms
��Ҫע����ǣ�ʯīϩ̼��Ԫ���������Ϸ���Hλ��Liԭ�����ȶ���λ�ã���������Lin�ž����ڲ���������ã�Liԭ�ӳ���λ�ûᷢ��ƫ�룬����������Liԭ��ʱ��Liԭ�ӻ���ͼ�������ٽ�̼��Ԫ���Ϸ����ž���Liԭ�ӵ��Ϸ����Ӷ�ʹʯīϩ�������������ܳ���ʯī���������γ��֦��������Liԭ������n�����࣬��������������ž�����ײ��Liԭ�ӵ�ʯīϩƽ���ƽ������h����hֵ��dLi-Cֵ��������������������ƣ�dLi-Cֵ��������Ƚ��ͺ������ȶ������ƣ������Lin�ž�����ʯīϩ֮��������������
3.2 ���ӽṹ����
Ϊ�˷���ʯīϩ����Lin�ž���ĵ��ת���������ʯīϩ�����������Liԭ�ӡ�Li4��Li7��Li9�ž������������ͽ��е��ӽṹ�����������ܶȲ�ַ��������ͼ5��ʾ�����е���ܶȲ��ͨ������=��[Lin-G]-��[nLi]-��[G]����õ�����[Lin-G]����[nLi]����[G]�ֱ����Lin-G��n��Liԭ�Ӻ�ʯīϩ�����ĵ����ܶȣ�ͼ�к�ɫ����ΪLiԭ��ʧȥ�����γɵ�ʧ����������ɫ����Ϊʯīϩ�����Lin�ž����ڲ��ĵõ�������
��ͼ5��֪����ʯīϩ�����������Liԭ�ӣ�Liԭ��ʧȥ�������ӣ���ת�Ƶ�ʯīϩ�����ٽ��ķ����й������ʯīϩ����������Եĵ��Ӿۼ�����ʯīϩ�������Li4�ž���ʱ���ž����������ʧ����״̬���ֵ���ת����ʯīϩ�����ٽ��ķ����й�����ٲ��ֵ��Ӿۼ���Li4�ž��������ڲ�����ʯīϩ�������Li7��Li9�ž���ʱ���ž�������Ҳ����ʧ����״̬�������ž����ڲ����־����ʧ��������͵õ�������Liԭ��֮�������ø����ӡ�

ͼ5 ʯīϩ�������Liԭ�ӻ�Lin�ž���ĵ����ܶȲ��ͼ(ͼ������ΪLiԭ��Mulliken����)
Fig. 5 Three-dimensional charge density difference of Li atom or Lin aggregate on graphene (The data in the figure are Mulliken population of Li atom)
Mulliken���ӷ���������ʯīϩ�����������Liԭ�ӡ�Li4��Li7��Li9�ž��������������У�Liԭ��ƽ��Mulliken���ӷֱ�Ϊ1.15e��0.58e��0.58e��0.40e��ʯīϩ�����ٽ���Cԭ��ƽ��Mulliken����Ϊ-0.12~-0.13e�����������Liԭ������n�����࣬Lin�ž�����ʯīϩת�Ƶ�ƽ����������١�
Ϊ�˶����о�ʯīϩ����Lin�ž����ڲ��Լ���ʯīϩ�ijɼ������������Ӿ����ܶȺ���(ELF)�Ե��ӽ��ж�������������ͼ6��ʾ������ELF�ķ�ΧΪ0��1����ELFΪ1ʱ����Ϊ��ǿ�Ĺ��ۼ�����ELFΪ0.5ʱ����Ϊ����������ELFΪ0��0.5ʱ���ֳ���ǿ�����Ӽ���
��ͼ6��֪����ʯīϩ�����������Liԭ��ʱ��Liԭ����ʯīϩ֮���γɵ�Li-C��Ϊ���Ӽ�����ʯīϩ�������Li4�ž���ʱ���ž���ײ��Liԭ����ʯīϩ֮���Li-C���Լ��ײ�Liԭ��֮���Li-Li����Ϊ���Ӽ����ž��嶥����ײ���Liԭ�ӹ������ӣ��γɵ�Li-Li��Ϊ���ۼ�����ʯīϩ�������Li7��Li9�ž���ʱ���ž�����ʯīϩ֮��ͬ��Ϊ���Ӽ���ϣ���Lin�ž���ijɼ��Ƚϸ��ӣ��Ŵصײ��Li-Li��ͨ��Ϊ���Ӽ���������Li-Li��ͨ��Ϊ���ۼ������ڲ��п���Ϊ���Ӽ���Ҳ����Ϊ���ۼ��������ž���Ĺ����Լ����ת���йء����Liԭ��ƽ��Mulliken���ӷ���������Liԭ������n�����࣬Lin�ž�����ʯīϩת�Ƶĵ�������٣�����ʯīϩ��Liԭ���γ�Li-C���Ӽ�ǿ�������������ž�����ײ�Liԭ�ӵ�ʯīϩƽ���ƽ������h��dLi-Cֵ������ı仯����һ�¡�

ͼ6 ʯīϩ�������Liԭ�ӻ�Lin�ž����ELFͼ
Fig. 6 Electron localization function of Li atom or Lin aggregate on graphene
ʯīϩ�Լ������������Liԭ�ӡ�Li4��Li7��Li9�ž����̬�ܶ�ͼ��ͼ7��ʾ������ʯīϩ��-20��-10 eVΪʯīϩsp2�ӻ��γɵġ�����-10��0 eVΪʯīϩ�Ħй����0��5 eVΪʯīϩ�ķ����й������ʯīϩ�������Liԭ���Ժ�Liԭ�ӵ�2s����ע��ʯīϩ�ķ��������ʯīϩ��������ĵ���̬�����ߣ�ͬʱLiԭ�ӵ�2s�����Cԭ�ӵ�2p��������ӻ��������γɵ�Li-C��Ϊ���в��ֹ��ۼ����Ե����Ӽ�������Liԭ������n�����࣬�����ܼ�������ƶ��������ܼ�ʼ���е���̬��Lin-G��ϵ�Ľ����Ժ͵��ӵ���������ǿ��
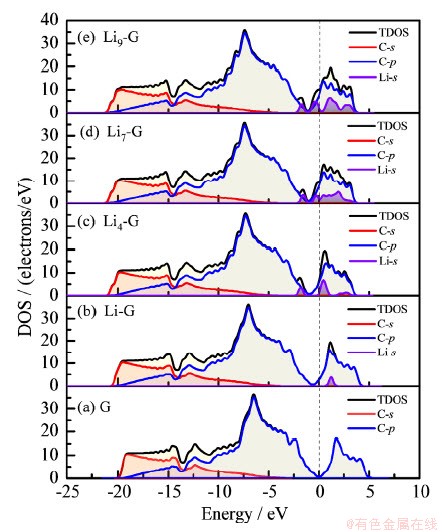
ͼ7 ʯīϩ���������Liԭ�ӻ�Lin�ž����̬�ܶ�ͼ
Fig. 7 Density of states of Li atom or Lin aggregate on graphene
3 ����
1��ʯīϩ�������Liԭ�ӻ�Lin(n<10)�ž��壬�����ܼ�������������n��2ʱ��Liԭ���Է�ɢ����ʽ�����ȳ�����ʯīϩ̼��Բ���������Ϸ�λ�ã���n��3ʱ��Liԭ�����Ⱦۼ������ž������ʽ������ʯīϩ���棬��ʵ�����п����γ��ȶ���Li4��Li7��Li9�ž��塣
2�����ӽṹ����������Liԭ�ӻ�Lin�ž���ĵ���ת�����ٽ�ʯīϩ�ķ����й����Liԭ�ӵ�2s�����Cԭ�ӵ�2p����������Ե��ӻ���������Liԭ����n�����࣬Lin-G��ϵ�ķ����ܼ���ʯīϩ�����й���ƶ�����ϵ�Ľ����Ժ͵��ӵ�������ǿ��
3��Lin�ž���ײ��Li-Li��ͨ��Ϊ���Ӽ���������Li-Li��ͨ��Ϊ���ۼ����ž�����ʯīϩ֮���Li-C��Ϊ���в��ֹ��ۼ����Ե����Ӽ�������Li-C��ǿ������Liԭ������n���������С��
REFERENCES
[1] Raccichini R, Varzi A, Passerini S, et al. The role of graphene for electrochemical energy storage[J]. Nature Materials, 2015, 14(3): 271-279.
[2] Dong Yanfeng, Wu ZhongShuai, Ren Wencai, et al. Graphene: a promising 2D material for electrochemical energy storage[J]. Sci Bull, 2017, 62(10): 724-740.
[3] ��, ���ܱ�, ��˼��, ��. ���ڵ�һ��ԭ��Sn-Li�Ͻ�Ƕ����ܺ͵������ܵļ�����Ԥ��[J]. �й���ɫ����ѧ��, 2017, 27(2): 282-288.
SHEN Ding, YANG Shao-bin, LI Si-nan, et al. Calculation and prediction of lithium insertion properties and elastic properties for Sn-Li alloy based on first-principle[J]. The Chinese Journal of Nonferrous Metals, 2017, 27(2): 282-288.
[4] ���侲, ���Ƚ, ��չ��, ��. ���ʯīϩ���Ʊ��봢������о�[J]. ��Դ����, 2020, 44(2): 153-155+222.
GUO Cui-jing, SHEN Jin-ran, LI Zhan-peng, et al. Synthesis and lithium storage performances of porous graphene[J]. Chinese Journal of Power Sources, 2020, 44(2): 153-155+222.
[5] ��ʫ��, ¹����, ����, ��. ʯīϩǶ﮵���������[J/OL]. ������ѧѧ��: 1-6 [2021-08-05].
http://kns.cnki.net/kcms/detail/11.1892.O6.20200317.1619.006.html.
Shi Yi-tang, Gao Tian-lu, Yi Su, et al. Raman mapping of lithiation process on graphene[J/OL]. Acta Physico-Chimica Sinica, 2021.
[6] ��ӱ. Li��ʯīϩ����������Ǩ�Ƶĵ�һ��ԭ���о�[J]. ���ϵ���, 2019, 33(S2): 43-47.
JIA Ying. First principle calculations on adsorption and diffusion behavior of Li on graphene surface[J]. Materials Reports, 2019, 33(S2): 43-47.
[7] Zhong Kehua, Yang Yanmin, Xu Guigui, et al. An ab initio and kinetic monte carlo simulation study of lithium ion diffusion on graphene[J]. Materials, 2017, 10(7): 1-17.
[8] Yildirim H, Kinaci A, Zhao Z J, et al. First-principles analysis of defect-mediated Li adsorption on graphene[J]. ACS applied materials & interfaces, 2014, 6(23): 21141�C21150.
[9] Liu Yuanyue, Artyukhov Vasilii I, Liu Mingjie, et al. Feasibility of lithium storage on graphene and its derivatives[J]. J Phys Chem Lett, 2013, 4(10): 1737-1742.
[10] Rani B, Jindal V K, Dharamvir K. Energetics of a Li atom adsorbed on B/N doped graphene with monovacancy[J]. Journal of Solid State Chemistry, 2016: 67-75.
[11] Wonsang Koh, Hye Sook Moon, Seung Geol Lee, et al. A first-principles study of lithium adsorption on a graphene-fullerene nanohybrid system[J]. Chem Phys Chem, 2015, 16(4): 789-795.
[12] David Adekoya, Zhang Shanqing, Marlies Hankel. 1D/2D C3N4/graphene composite as a preferred anode material for lithium ion batteries: importance of heterostructure design via DFT computation[J]. ACS applied materials & interfaces, 2020, 12(23): 25875�C25883.
[13] ����, ��С��, �շ�Զ, ��. ʯīϩ/̿���ӻ�����: ���͡���Ч����ӵ�ض�Ԫ�����[J]. ����̿����, 2015, 30(2): 128-132.
LI Yong, LU Xiao-hui, SU Fang-yuan, et al. A graphene/carbon black hybrid material: a novel binary conductive additive for lithium-ion batteries[J]. New Carbon Materials, 2015, 30(2): 128-132.
[14] Wei Xufang, Guan Yibiao, Zheng Xiaohui, et al. Improvement on high rate performance of LiFePO4 cathodes using graphene as a conductive agent[J]. Applied Surface Science, 2018, 440: 748-754.
[15] VANDERBILT, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990, 41, 7892-7895
[16] Segall M D, Lindan P J D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[17] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192.
[18] Chang C, Yin S, Xu J. Exploring high-energy and mechanically robust anode materials based on doped graphene for lithium-ion batteries: a first-principles study[J]. RSC Advances, 2020, 10(23):13662-13668.
[19] Lee E, Persson K A. Li absorption and intercalation in single layer graphene and few layer graphene by first principles[J]. Nano Letters, 2012, 12(9): 4624-4628.
Deposition behavior of Lin (n<10) aggregates on graphene based on first-principles calculations
SHEN Ding1,2, WANG Lai-gui2, TANG Shu-wei1, GAO Si-da1, SUN Wen1, Dong Wei#1, YANG Shao-bin#1
(1 College of Materials Science and Engineering, Liaoning Technical University, Fuxin 123000;
2 School of Mechanics and Engineering, Liaoning Technical University, Fuxin 123000, China)
Abstract: Deposition behavior of Li atoms or aggregates Lin (n<10) on graphene negative electrodes for lithium-ion batteries was investigated by the first principles calculations based on density functional theory. The calculation result shows that Li atom preferentially deposits above the center of the carbon six-membered ring of graphene in a dispersed form when n��2. As the number of Li atoms increases, Li atoms preferentially aggregate to reunite and deposit on graphene when n��3. It is possible to form Li4, Li7 and Li9 stable agglomerates in the charging process of the lithium ion battery, which indicates that the maximum lithium storage capacity of graphene may exceed that of graphite. However, the lithium dendrites would be formed easily. The electronic structure analysis shows that the electrons of Li atom or Lin aggregates transfer to the anti-bonded �� orbital of graphene, and the 2s orbital of Li atom and the 2p orbital of C atom are obviously hybridized. The Fermi level of the system moves to the graphene anti-bond �� orbital as the number of Li atoms n increases, which led to the result that the metallicity and electronic conductivity increase. The Li-Li bond at the bottom of the Lin agglomerate is usually an ionic bond and the outermost Li-Li bond is usually a covalent bond. The Li-C bond between the Lin agglomerate and graphene is an ionic bond with partial covalent bond property and the strength of the Li-C bond gradually decreases as the number of Li atoms n increases.
Key words: lithium ion battery, graphene, Lin agglomerates, first-principles, deposition behavior
Foundation item: Projects (51874167, 21808095, 51774175) supported by the National Natural Science Foundation of China; Projects (2018M641707) supported by the China Postdoctoral Science Foundation Funded; Projects (LNTU20TD-09) supported by the Discipline Innovation Team of Liaoning Technical University.
Corresponding author: YANG Shao-bin; Tel: 0418-3352741; E-mail��lgdysb@163.com
������Ŀ��������Ȼ��ѧ����(51874167��21808095��51774175)����ʿ�����ϻ���(2018M641707)���������̼�����ѧѧ�ƴ����Ŷ�������Ŀ(LNTU20TD-09)
�ո����ڣ�2020��07��31�������ڣ�2020��11��13
ͨѶ���ߣ����ܱ��ڣ���ʿ���绰��0418-3352741�������ʼ���lgdysb@163.com
ժ Ҫ�����û����ܶȷ����ĵ�һ��ԭ���������о�������ӵ��ʯīϩ����Liԭ�ӻ��ž���Lin(n<10)�ij�����Ϊ�������������Liԭ����n��2ʱ��Liԭ�������Է�ɢ����ʽ������ʯīϩ̼��Ԫ�������Ϸ�����n��3ʱ��Liԭ���������ž������ʽ������ʯīϩ���棬�����γ��ȶ���Li4��Li7��Li9�ž��塣�����ʯīϩ�������������ܳ���ʯī���������γ��֦����Liԭ�ӵĵ���ת�����ٽ�ʯīϩ�ķ����й����Liԭ�ӵ�2s�����Cԭ�ӵ�2p����������Ե��ӻ���������n������ϵ�ķ����ܼ���ʯīϩ�����й���ƶ��������Ժ͵��ӵ�������ǿ��Lin�ž���ײ��Li-Li��ͨ��Ϊ���Ӽ���������Li-Li��ͨ��Ϊ���ۼ���Li-C��Ϊ���в��ֹ��ۼ����Ե����Ӽ�������Li-C��ǿ������n����������С��
[4] ���侲, ���Ƚ, ��չ��, ��. ���ʯīϩ���Ʊ��봢������о�[J]. ��Դ����, 2020, 44(2): 153-155+222.
[5] ��ʫ��, ¹����, ����, ��. ʯīϩǶ﮵���������[J/OL]. ������ѧѧ��: 1-6 [2021-08-05].
http://kns.cnki.net/kcms/detail/11.1892.O6.20200317.1619.006.html.
[6] ��ӱ. Li��ʯīϩ����������Ǩ�Ƶĵ�һ��ԭ���о�[J]. ���ϵ���, 2019, 33(S2): 43-47.
