
���±��: 1004-0609(2005)03-0391-06
�������VHx(x=0, 1, 2)���ӽṹ��DV-X���о�
�� ��1, 2, ������1, ������2, ����ƽ1, �²���1
(1. �����ѧ ���Ͽ�ѧ�빤��ѧԺ, ���� 400044;
2. ����ʦ����ѧ ��ѧѧԺ, ���� 400047)
ժ Ҫ��
���õ����Ǣ��ɢ���X��(SCC-DV-X��)���������˷����������з��ⷴӦǰ�����⻯��(VHx, x=0, 1, 2)�ĵ��ӽṹ�� �Ծ���ɡ� �����͵����ܶȲ�ķ�������: VH��VH2��V��H��֮��������������й����Ե�����á� VH2��V��H֮��ļ�����VH��V��H֮��ļ���С, ˵��VH2�е�H�����ͷų����� ���ܼ��ṹ�� ̬�ܶȺͼ۹���������ķ�������, �⻯��VH����V��4s�����H��1s������óɼ�; �⻯��VH2����V��4s�� 3d�����H��1s������óɼ�; VH��VH2�ķ����ܼ���, ˵��VH���ȶ��� ������VH2�е��ⲻ��ȫ���ų���ԭ��
�ؼ���: ���⻯��; ���ӽṹ; SCC-DV-X������
��ͼ�����: O641.121 ���ױ�ʶ��: A
Quantum chemical DV-X�� study on electronic structure of hydrogen storage materials VHx(x=0, 1, 2)
LI Rong1, 2, ZHOU Shang-qi1, LIANG Guo-ming2, LIU Shou-ping1, CHEN Chang-guo1
(1. College of Materials Science and Engineering, Chongqing University, Chongqing 400044, China;
2. College of Chemistry, Chongqing Normal University, Chongqing 400047, China)
Abstract: The electronic structure of metallic V before and after reaction of V��H and its hydrides(VHx, x=0, 1, 2) in vanadium base solid solution was calculated by a quantum chemical method(SCC-DV-X��). By analyzing net charge, bond level and electron density difference, the following results are obtained: there exists interaction between V��H bands interaction which has not only ionic but also covalent property in hydride VH and VH2, bond level between V��H in hydrideVH2 is weaker than that in VH, which proves that H in VH2 is released more easily than in VH. By analyzing the energy level structure and electron density of states and the electron occupation numbers in valence orbits, the result show that the former V 4s-H 1s interaction is bonding-type, and also the latter V 3d, 4s-H 1s interaction is bonding-type. Fermi energy level in VH is weaker than that in VH2, which proves that VH is more stabilization, and explains the reason why H2 is not all released.
Key words: vanadium hydride; electronic structure; SCC-DV-X�� method
���ڴ�ͳ��AB5�� AB2��AB��������ϵ�������������2%(��������), ���������������ȼ�ϵ���ϵ�Ӧ��, �ʶ��ڸ�������������ϵ��о����ܹ�ע�� ���������������Ϊһ�����͵��������, ���п����������� �����⻯���е���ɢ�ٶȽϿ���ŵ�, ���й����ķ�չǰ���� Ŀǰ�Է�����������������ϵ��о�������Ҫ���������õ�V3TiNi0.56Mx(x=0.046~0.24, MΪAl�� Si�� Mn�� Fe�� Co�� Zr����)�ȡ� ͨ���Ż��Ͻ����ṹ����ɽ�һ�����V���������Ͳ��ϵ��ۺϵ绯ѧ����, �Լ��о������۸������V��V�Ͻ�ԭ�����Խ���������ϳɱ�, ��ĿǰV����������������ϵ�������Ҫ�о����� ��������������ʱ������VH��VH2���ֲ�ͬ���͵��⻯��, ��P-C-T�����ϳ���������Ӧ��ѹ��ƽ̨�� V��������Ͻ������������(Լ3.8%)���ص�, ����̬��������������20����70����ѽ���ϵͳ�о��� �о�����, ��������V���������⻯���ɵ�VH���⻯������ȶ���������, ����ƽ̨��Ӧ��VH2��VH+H��������������, �Ͻ�Ŀ����������ɴ�1.9%����, �Դ���AB5�� AB2��������ϡ� ��������������������������V�ĺ������, ��������������Ͻ�����ķ�Ӧ��Ҫ��V��H2�ķ�Ӧ, ��˶Է��ⷴӦ���о��dz���Ҫ�� ��һЩѧ�߶Դ˽�����ʵ���о�[1-5]��
���������о����ⷴӦ������ѧ������������[6]�þ����ܶȷ������Ʒ����Խ���������ͬԭ�ӱȵķ��⻯������˽ṹ�Ż�������������; Matumura��[7]��DV-X�������о��˺����ֺϽ�Ԫ�ص�VH2��V2H�ĵ��ӽṹ; Yukawa��[8, 9]��DV-X�������о���LaNi5�� TiFe�� ZrMn2�� Mg2Ni��V���⻯��ĵ��ӽṹ, �����������⻯����ȶ��Ե�ԭ�Ӽ�������ںܴ�̶���ȡ�������⻯�����о���ṹ�ݱ�ķ�ʽ�� ��V��������VH, �Լ�VH��������VH2������, �⻯��ĵ��ӽṹ���������ܵĹ�ϵ��δ�������� ��������Ӧ�����ӻ�ѧDV-X�������о�V��H��Ӧ����VH�� VH2������ԭ�ӵľ���ɡ� �ܼ��ṹ�� ̬�ܶȡ� ������ �����ܶȵı仯, ̽������������ϵ���������֮��Ĺ�ϵ��
1 ģ���뷽��
1.1 �ṹģ��
����Ӧ�á�����ԭ�ӷ���(seed atoms)[10], ѡ�� Vԭ����Ϊ����ԭ��, ���ϵؽ������ԭ�Ӱ���Զ��˳���������Ŵ��С� ���ľ���ṹΪ������������(bcc), ѡȡ27��������91��ԭ����Ϊ����ģ��, ��������a=0.3029nm[7], �ռ�ȺΪTd�� �����������, �侧��ṹ������ת�䡣 ����������(��a+������)�ں������ܵ�ʱ�γ������ķ�����, ������Ϊ������Ťת�������ķ�����(bct), ���dz���ļ�������, ѡȡ59��Vԭ�Ӻ�56��Hԭ����Ϊ����ģ��, �侧������a=0.3001nm, c=0.3395nm[7], �ռ�ȺΪD2h; VH2�ľ���ṹΪ������������(fcc), ÿ��Hԭ��ռ�ݾ����е�8cλ��, ����������������ġ� ѡȡ63��Vԭ�Ӻ�64��Hԭ����Ϊ����ģ��, ��������a=0.4271nm[7], �ռ�ȺΪTd�� VHx(x=0, 1, 2)��ԭ�Ӵ�ģ����ͼ1��ʾ, ģ���е����ֱ�ʾ�ռ�Ⱥ��ԭ�ӵ�����(����������е����������ͬ)��
1.2 ���㷽��
DV-X��(��ɢ���X��)�������ڷ�����۵�����Hamiltonian���:
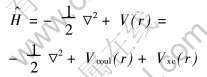
ʽ�� ������Vcoul(r)�Ƿ����и�ԭ�Ӻ˶Ե��ӵ������͵���֮����ų���, ��
![]()
Vxc(r)�ǵ��Ӽ�Ľ��ƽ�����:
![]()
ʽ�� ���ǽ�������, ȡֵ��Χͨ��Ϊ2/3�ܦ���1, ���ļ�����ȡ��=0.7�� ��LCAO-MO����, �����Ӳ�������ԭ�ӹ��չ��Ϊ ![]() , ϵ��Cki��ͨ����ֵ�����ڷ��̵õ���
, ϵ��Cki��ͨ����ֵ�����ڷ��̵õ���
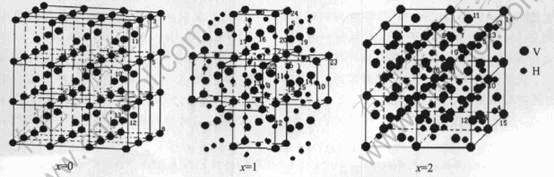
ͼ1 VHx(x=0, 1, 2)��ԭ�Ӵ�ģ��
Fig.1 Cluster modes of VHx(x=0, 1, 2)
DV-X������������Ľ�, Ŀǰ�ļ�������Ϊ��ԭ����420���� ��������55���� �ܵ�����3000��[11, 12]��
����ɢ���X��ԭ�Ӵؼ�����, ���ö���о�������������ģ�ͻ�����ȡ��ֵԭ�Ӷ�λ���(SSO)��, ��VΪ(1s2s2p)3s3p3d4s, HΪ1s2s (�����ڵ�ԭ�ӹ��Ϊ��о���), ��Ӱ뾶ΪV: 6.0�� H: 4.0, ��ΪV: -1.5�� H: -1.0�����塣 ȡ����ֲ�����Diophantus����, ÿ��ԭ����Χ500����, ��Ǣ��������������Ϊ10~5�� ���������Ʒ�[13]�������߽�ЧӦ��
1.3 ���ӽṹ����
Ҫ�о�VHx(x=0, 1, 2)�е��ӽṹ�ͻ�ѧ��������, ����Ҫ�о����ǵ��ܼ��ṹ�� ����̬�ܶȿ������ص���˹������������� ����Mulliken����������������֤���ӵ��ص��Լ�������ÿ��ԭ�ӵĴ�������� ���ϵ�������Ҫ�ɻ�ѧ������, ���ۼ������Ӽ���������Ҫ�Ļ�ѧ���� ���Ӽ��ڶ����ϺͿ�����������, ����ԭ��������ɵij˻�������, ��ԭ��֮������ƽ���ɷ��ȡ� ԭ�������ľ������ԭ��������ԭ�ӵļ�������ܲ���֮��IJ�ֵ�� ���ۼ���ǿ�ȿ�����ԭ��֮��Ĺ��ۼ�����ռ�ݵ�ԭ�ӹ��֮����ص������������� ���ۼ����ɵ��ӽṹ����, ��ӳ��ԭ��֮������Ƶijɼ��ص��̶�, ���ۼ���Խ��, ԭ�Ӽ乲�ۼ�Խǿ�� һ���˵, A��B���Ĺ��ۼ�����A��Bԭ��֮����ص�����P(A��B), �䶨��Ϊ[14]
![]()
ʽ�� ��i�ͦ�j�ֱ���A��Bԭ�ӵĵ�i��j��ԭ�ӹ��, CAni��CBnj�ֱ��Ǹ�ԭ�ӹ���ڷ��ӹ���е����ϵ��, ��n����ͱ鼰���е���ռ�ݵķ��ӹ����
2 ���������
2.1 �����
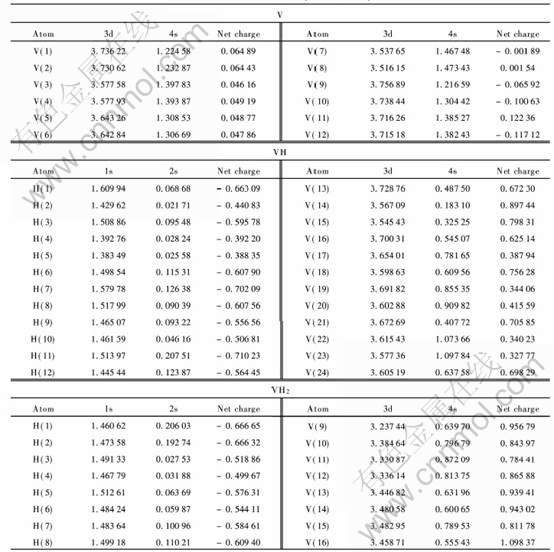
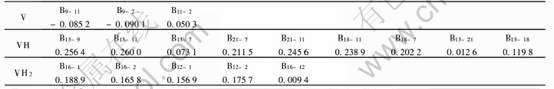
������ÿ��ԭ�ӵĴ���������1����, ��������ÿ��ԭ��������ɷdz�С, ���������ұ仯, ����V��Ϊ�������ʵ����۽������Ǻϡ� �������Ϊ�⻯��VH�Ժ�, Vԭ�����Ǵ�����, ��Hԭ�����Ǵ�����, ˵��V�����H��������Ԫ��; �⻯��VH�е�ɵ�ת����Ҫ�Ǵ�Vԭ��ת�Ƶ�Hԭ��, ���ڵ�ɵ�ת�Ƶ����������Ӽ�������, ����VH�з���֮���������á� Hԭ����Vԭ��֮����������Ҫ����������֮���ɵ�ת������, ת�Ƶĵ����Խ��, �����������Խǿ�� V�ľ������+0.32~+0.89��Χ֮��, ��VH��V�Ļ��ϼ�+1�����һ���IJ�ֵ; H�ľ������-0.44~-0.71֮��, ��VH��H�Ļ��ϼ�-1���Ҳ��һ���IJ�ֵ�� ����VH��V��H���ȴ��������Ե������, �ִ��ڹ����Ե�����á� ����һ�������Ϊ�⻯��VH2�Ժ�, ��VH��һ��, Vԭ�����Ǵ�����, ����ԭ�����Ǵ�����, ����VH2�е�ɵ�ת��Ҳ�Ǵ�Vԭ��ת�Ƶ�Hԭ�ӡ� ����VH2��, V�ľ������0.78~1.02֮��, ��VH2��V�����ۻ��ϼ�+2���ϴ� Hԭ�ӵľ����ֻ������С�ı仯, ��-0.47~-0.71֮��仯����-0.50~-0.67�� ��һ����Hԭ�ӵõ�����, һ����Hԭ��ʧȥ����, ����VH2��V��H��������üȴ��������Ե������, �ִ��ڹ����Ե�����á�
2.2 Mulliken������
��1������V��3d�� 4s�����H��1s�� 2s�����Mulliken������, ���Կ���, V+H��VH�Ĺ�����, V��4s����ϵĵ������Լ���, H��1s����ϵĵ�������������, V��3d�����H��2s����ϵļ������仯�dz�С�� ˵����V��4s����ϵĵ��������1s���ת��, Ҳ����˵, �⻯��VH��, V��H֮��������������V��4s�����H��1s����ɼ��� V��һ������ʱ, ����VH+H��VH2������, V��3d����ϵĵ��������ڼ��١� V��4s����ϵĵ���һ���ּ���, ��һ����ȴ����, ��������V��3d�����4s����������ӻ��� Hԭ�ӵ�1s�����2s����ϵĵ�����û�����ԵĹ���, �����������м��١� ˵���⻯��VH2��, V��H֮��������������V��4s�� 3d�����H��1s���֮��ɼ���
2.3 �ܼ��ṹ�͵���̬�ܶ�
ͼ2�ֱ������V�� VH�� VH2���ܼ��ṹ�� �ܵ���̬�ܶ���H��1s�� V��3d�� 4s����ķ�̬�ܶȡ� ��ͼ�п������Կ���, ��V�� VH�� VH2�ж���V��3d����������ֲ���Լ���, ���ܵ���̬�ܶȵĹ������, ��H��1s��V��4sȴ�����������ֲ��dz���ɢ������� �����ܼ�Efλ��V��3d���� ��V�� VH�� VH2�ж���Ef��Χ��̬�ܶ� ���, ˵�����ǽ���ʧ���ӡ� ͼ2(b)��, �����
��1 VHx(x=0, 1, 2)��ԭ�ӵľ���ɺͼ۹���ĵ��Ӽ�����
Table 1 Net charge on each atoms and electron occupation numbers in
valence orbits of VHx(x=0, 1, 2)
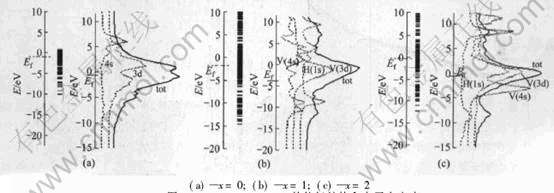
ͼ2 VHx(x=0, 1, 2)���ܼ��ṹ�͵���̬�ܶ�
Fig.2 Energy level structure and electron density of states for VHx(x=0, 1, 2)
��λ��Ef���µ�H��1s�����V��4s��������ص�, ��������֮���гɼ�����; λ��Ef���ϵ�1s�����V��4s���Ҳ�в����ص�, ��������֮�����з������á� �Ӷ�˵��VH������, Vԭ�Ӻ�Hԭ��֮������ǿ�ҵ�����á� ͼ2(c)��, �������λ��Ef���µ�H��1s�����V��3d��������ص�, ˵��H��1s�����V��3d���֮���гɼ�����; λ��Ef���ϵ�H��1s�����V��4s��������ص�, ˵��1s�����V��4s���֮���з������á� ����ǰ��Ľ���һ�¡� �Ƚ�ͼ2(b)��2(c)�ɼ�, ��VH+H��VH2�ı仯������, H��1s�����̬�ܶ����Ե�������������ƶ�, ��V��3d��4s�����̬�ܶ���������������ƶ���һЩ��
��ΪEf���⻯����ȶ��������е���ϵ, EfԽ��, ����ѹԽС, �⻯���ȶ���Խ��[15]�� VH�� VH2�ķ����ܼ�Ef�������ֱ�Ϊ: -1.79eV�� -0.12eV�� ��VH�ķ����ܼ���VH2�ķ����ܼ���, ����VH��VH2���ȶ��� ��ΪEf���⻯����ȶ��������е���ϵ, EfԽ��, ����ѹԽС, �⻯���ȶ���Խ��[15]�� VH�� VH2�ķ����ܼ�Ef �������ֱ�Ϊ: -1.79eV�� -0.12eV�� ��VH�ķ����ܼ���VH2�ķ����ܼ���, ����VH��VH2���ȶ�, VH2���ֽ�����VH��H; ��V�ķ�����Ϊ-1.12eV, VH�ķ����ܱ�V�ķ�����Ҳ��, VH�ٷֽ�ΪV��H���ѡ� �����⻯��VH2�е��ⲻ����ȫ���ͷų�����
2.4 ԭ��֮��ļ���
��2�����ˡ����ӡ�(����Բ��)ԭ��(Vԭ��)����Χ���в�ͬ���͵�Vԭ�ӡ� Hԭ�ӵļ����� ��VH��VH2��, ����V֮��ļ������Ǹ�ֵ���ӽ�����, ˵��V-V֮��Ĺ���������ý�С; �෴, Hԭ�Ӻ�Vԭ��֮��ļ����ܴ���Ҷ�����ֵ, ��ͱ���V��H��֮���ǹ����Ե�����á� ������VH��, V��H֮��ļ��������, V(13)��H(9)��V(13)��H(7)���ϴ�, ��������С��V(13)��H(7)��������V��H֮�������ø�ƫ�������ԡ� ����VH2��, V��H֮��ļ�������VH��V��H֮��ļ���С, ��������֮�������ö�ƫ����, ����V��H����ǿ�Ƚ���, ����VH2�е�H�����ͷų����� ����ǰ�澻��ɵ����ۺͰ����ֵĵ縺�����۽��������Ǻϵġ� ��ΪV�ĵ縺��Ϊ1.6, ��H�ĵ縺��Ϊ2.1, ���ǵĵ縺��֮��С��1.7, ����V��H֮��������ƫ���ԡ�
2.5 �����ܶȲ�
Ϊ�˽�һ���˽⡰���ӡ�ԭ��(Vԭ��)����Χ���в�ͬ���͵�Vԭ�ӡ� Hԭ�ӵ������, ������������V�� VH�� VH2�о��������ӡ�ԭ�ӵ�(110)��ĵ���ܶȲ�(ͼ3)�� ����ܶȲ��ʾԭ��֮��ĵ����ص������ ��ͼ1���Կ���, ��VH��, һ����ԭ���뷰ԭ��֮��ĵ������ص��϶�, ˵��V��H��֮����Ҫ�ǹ����������; һ����ԭ�� �뷰ԭ��֮��ĵ������ص�����, ����V��H��֮����Ҫ������������á� ����VH2��, ���е���ԭ���뷰ԭ��֮��ĵ������ص����Ƚ϶�, ����V��H��֮��Ĺ���������ý�ǿ, ����ǰ��Ľ������Ǻϡ�
��2 �����VHx(x=0, 1, 2)�Ĺ��ۼ�����
Table 2 Calculated covalence levels of VHx(x=0, 1, 2)
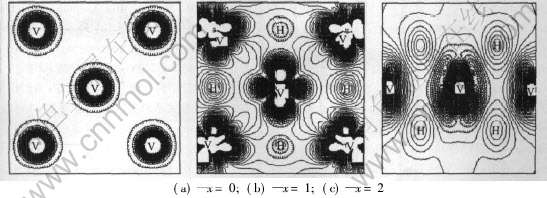
ͼ3 VHx(x=0, 1, 2)��(110)��ĵ����ܶȲ�
Fig.3 Electron density difference of VHx(x=0, 1, 2) on plane (110)
3 ����
���õ����Ǣ��ɢ���X��(SCC-DV-X��)���������˷������⻯��(VHx, x=0, 1, 2)�ĵ��ӽṹ, �������:1) �⻯��VH�� VH2��V��H��֮��������������й����Ե������; 2) VH2��V��H֮��ļ�����VH��V��H֮��ļ���С, ˵��VH2�е�H�����ͷų���; 3) �⻯��VH��V��4s�����H��1s������óɼ�; �⻯��VH2��V��4s�� 3d�����H��1s������гɼ��������з�������; 4) �⻯��VH��VH2�ķ����ܼ���, ˵��VH���ȶ��� ͨ�����ϼ���, ������VH2�е��ⲻ��ȫ���ų���ԭ��
REFERENCES
[1]Budylkin N, Voloschin L, Mironova E, et al. Hydrogen isotopes mobility and trapping in V-Cr-Ti alloys[J]. Journal of Alloys and Compounds, 1996, 233-237: 1160-1162.
[2]Dug R, Nowicka E, Wolfram Z. Surface phenomena in the process of vanadium hydride formation[J]. Journal of Alloys and Compounds, 1997, 253-254: 496-499.
[3]Yamashita S, Komiya D, Yukawa K. Compositional dependence of phase stability of �� phase formed in vanadium alloys[J]. Journal of Alloys and Compounds, 2004, 364: 137-140.
[4]Buzlukov A L, Skripov A V. Nuclear magnetic resonance study of hydrogen motion in C15-type TaV2Hx (x�� 0.18) [J]. Journal of Alloys and Compounds, 2004, 366: 61-66.
[5]Tsukahara M, Takahashi K, Isomura T. Influence of oxygen on hydrogen storage and electrode properties for micro-designed V-based battery alloys[J]. Journal of Alloys and Compounds, 1998, 265: 257-263.
[6]������, ������. �����������⻯��ľ���ṹģ��[J]. ԭ���ܿ�ѧ����, 2000, 34(5): 469-472.
PENG Shu-ming, ZHAO Peng-ji. Simulation on crystal structures of metallic V and its hydrides[J]. Atomic Energy Science and Technology, 2000, 34(5): 469-472.
[7]Matumura T, Yukawa H, Morinaga M. Alloying effects on the electronic structures of VH2 and V2H[J]. Journal of Alloys and Compounds, 1999, 284: 82-88.
[8]Yukawa H, Matsumura T, Morinaga M, et al. Chemical bond state and hydride stability of hydrogen storage alloys[J]. Journal of Alloys and Compounds, 1999, 293-295: 227-230.
[9]Yukawa H, Takagi M, Teshima A, et al. Alloying effects on the stability of vanadium hydrides[J]. Journal of Alloys and Compounds, 2002, 330-332: 105-109.
[10]ͯ����, ��ĵ, ��ѧ��. PbWO4���ӽṹ���ܶȷ�������[J]. ����ѧ��, 2000, 49(8): 1545-1548.
TONG Hong-yong, GU Mu, TANG Xue-feng. Theoretic calculation on the electronic structure of PbWO4 crystals[J]. Acta Phys, 2000, 49(8): 1545-1548.
[11]����. ����ӵ����ϵ�������ϵ��ӽṹ�����ӻ�ѧ�о�[D]. ����: �����ѧ, 2002.
LI Rong. The Quantum Chemical DV-X�� Study on the Electronic Structure of Lithium-Manganese-Oxides Electrode Material for Lithium Ion Battery[D]. Chongqing: Chongqing University, 2002.
[12]����, �²���, ������, ��. ����ӵ����������LixMn2O4���ӽṹ�����ӻ�ѧDV-X���о�[J]. �й���ɫ����ѧ��, 2004, 14(5): 865-870.
LI Rong, CHEN Chang-guo, LIANG Guo-ming, et al. Quantum chemical DV-X�� study on electronic structure of electrode material LixMn2O4 for lithium ion battery[J]. The Chinese Journal of Nonferrous Metals, 2004, 14(5): 865-870.
[13]��ع��, ���, ף�̿�. X�����������ۺ�Ӧ��[M]. ����: ��ѧ������, 1987.
PAN Yu-gang, LI Jun-qing, ZHU Ji-kang. Theory and Practice of Discrete Variational Method[M]. Beijing: Science Press, 1987.
[14]������, ������, ������, ��. S-M(M=Al, Co)���ϲ��ӵ�LiMn2O4�ṹ�ȶ���[J]. ������ѧѧ��, 2004, 20(3): 233-236.
ZHAO Shi-xi, MIN Xin-min, LIU Han-xing, et al. The structure stability of S-M(M=Al, Co) co-doped spinel LiMn2O4 cathode materials[J]. Acta Phys Chim Sin, 2004, 20(3): 233-236.
[15]����, ����, Ҷ��, ��. RENi5���ӽṹ���������ܹ�ϵ�о�[J]. ��ѧͨ��, 1995, 40(24): 2234-2236.
CHEN Ning, LIN Qing, YE Wen, et al. Relationship of electronic structure of RENi5 and hydrogen storage properties[J]. Chin Sci Bulletin, 1995, 40(24): 2234-2236.
(�༭Ԭ��ǰ)
������Ŀ: ��������Ȼ��ѧ����������Ŀ(CSTC2004BB4169); ����ʦ����ѧ��ʿ���л���������Ŀ
�ո�����: 2004-07-20; ������: 2004-12-13
�����: �� ��(1970-), ��, ��ʦ, ��ʿ.
ͨѶ����: �� ��, ��ʿ; �绰: 023-65362702; E-mail: Rongli258@hotmail.com
[6]������, ������. �����������⻯��ľ���ṹģ��[J]. ԭ���ܿ�ѧ����, 2000, 34(5): 469-472.
[10]ͯ����, ��ĵ, ��ѧ��. PbWO4���ӽṹ���ܶȷ�������[J]. ����ѧ��, 2000, 49(8): 1545-1548.
[11]����. ����ӵ����ϵ�������ϵ��ӽṹ�����ӻ�ѧ�о�[D]. ����: �����ѧ, 2002.
[13]��ع��, ���, ף�̿�. X�����������ۺ�Ӧ��[M]. ����: ��ѧ������, 1987.
[15]����, ����, Ҷ��, ��. RENi5���ӽṹ���������ܹ�ϵ�о�[J]. ��ѧͨ��, 1995, 40(24): 2234-2236.
