
��������ֵ��ͨ�ⷽ�����л���������繹��
����1, 2��֣��1������ϼ1������1������1���ܴ���1������1
(1. ���ִ�ѧ �������ѧ�뼼��ѧԺ������ ������130012��
2. ������ѧ �������ѧ����ѧԺ������ ������132021)
ժ Ҫ��
������ַ���ģ�ͻ����ϣ���������ֵ�ֽⷽ�����������㡣���㷨����ͨ������ֵ�ֽ���ؽ�õ���������ս��������ͨ������õ�ͨ���ṩ��ѡ�⼯�ķ�����Ϊ�ַ���ģ���㷨��������ʱ�����߽�����ȡ��������㷨��������ͳ�ַ���ģ���㷨���жԱȣ���֤������������㷨��Ч�ʽϸߡ�
�ؼ��ʣ�
����ֵ�ֽ����ַ���ģ�������������������ѡ�⼯��ͨ�����ޱ����������ѧ��ģ��
��ͼ����ţ�TP18 ���ױ�־�룺A ���±�ţ�1672-7207(2012)04-1377-05
Construction of gene regulatory network based on singular value decomposition for solution set
SHEN Wei1, 2, ZHENG Ming1, LIU Gui-xia1, XING Chong1, WU Jia-nan1, ZHOU Chun-guang1, ZHOU You1
(1. College of Computer Science and Technology, Jilin University, Changchun 130012, China;
2. College of Computer Science and Technology, Beihua University, Jilin 132021, China)
Abstract: The singular value decomposition method was introduced to calculate the network based on the improved decomposed weighted value differential equation model. Differently from the existing singular value decomposition model used for getting final network, the singular value decomposition method was used to get the solution set. The algorithm for simulation data and the real data were tested and compared with other algorithms. The results show that the proposed algorithm has high precision.
Key words: singular value decomposition; differential equation model; gene regulatory network; solution set; general solution; scale-free network; mathematical model
���Ż���������(cDNA)�����м����Ľ����������Ѿ��ܹ������������鷶Χ�ڽ��л����������Ĺ�������Щͨ��ʵ���õ������ṩ����һЩ�ض�ʱ�����ֲ�ͬ�����������»������ġ����м�ֵ��������������Щ�������о����ܴӻ����ʱ�����ֵ���ƶϻ���������硣������ˣ���Ҫ�Ļ�����������Ǵ����ڻ������еij�ǧ����Ļ��������ɵ����磬���临�ӡ����ԣ���ϵͳ����ѧ��һ�����ս�Ĺ������ǴӸ�������������ʱ���������ƶϳ�����������硣�ڿɹ۲�ϵͳ�ʹ��㷨�л�õ�����֮���Ǵ��ڲ���ģ����ֲ��쵼�����㷨��ȷ�ȵIJ��죬Ŀǰ�����ṩһ���߶Ⱦ�ȷ���㷨��ʵ�ֶ�����ʵ�����ݵ������ƶϡ�������ˣ��о�����ȷ�������ƶ�ģ����Ȼ��������Ϣѧ���о��ȵ㡣������������Ӧ���ڻ�����������ƶ��ϣ���Щ����������������[1]����Ҷ˹����[2-3]���ַ���ģ��[4]�ȡ����������ṩ��һ������ṹ��ϵ�ĸ���㷽��������һ���ģ�Ӵ��������ɰ���ǧ�Ļ����ǣ������нڵ��״ֻ̬�п���2��״̬����Ҷ˹������һ���ܹ����������ϵ�ĸ���ģ�ͣ�����ģ����ÿ���߶�����1�����ʣ���Ҷ˹�����ͼ�ι��ɵ���1��������ͼ���ڵ�һ�������ʮ���ڵ㣬��ģ���С��ַ���ģ������һ�ָ߶Ⱦ�ȷ�����繹��ģ�ͣ��Ա仯�ʵ���ʽ��ʾ�����������Ӱ���ϵ����һ����̬�����顣 ���ַ�ʽ���ɵ���������ֻ������������һ����20�����ڣ������ɵ���������ѧ���ȷ������ģС�������������۲������ַ�������Ȼ���ھ��Ȳ��ߵ����⣬��ˣ���Ҫһ�����õķ�������һ���Ľ��ַ���ģ�͡��Ľ�����֮һ�Ǹı䷽�̵���ʽ����ѷ�����ij���������зֽ��ı䣬�����������⻯Ϊ��������[5]�ȣ������ֲ����ڱ����ϲ�û�иı侫�ȡ���һ�ָĽ���������������ֵ�ֽ�(SVD)[6]�ķ���������������аѷ������б������������ɵľ������ת�þ�����зֽ⣬֮����о������㲢�õ���������ֵ�ֽ����Ľ⼴Ȩֵ������Ϊ�������������ڶ�������ʵ�����������£�������Ľ���������Ψһ��������ֵ�ֽ��������Ωһ�⣬���������������⼯�в��������Ž⡣���������⣬����������ͨ������ֵ�ֽ���õ��ؽ�����������������ֵ�ֽ�����ͨ�⼴��ļ��ϣ��������ͨ����Ϊһ��ȡֵ��Χ����ϸĽ���ʽ���ַ���ģ�Ͳ������Ż��㷨�����Ż������Ȩֵ���ó���������ս����
1 �㷨����ѧ���
�����ƶ�����ķ������Է�Ϊ2�������ȣ�Ҫʹ������ֵ�ֽⷽ��������һ����ļ��ϡ�����������ɷ��Ϸ�������ɵĽ⼯����ɵģ������ģ�ܴ�һ���ṩͨ�⡣Ȼ����������ϻ���½���ģ�͵��Ż���ʹ�����Իع���������ַ��̵���⣬�����յõ��������硣
Ϊ�˼�������̬ϵͳ���߽ӽ���̬��ϵͳ���з����������������ѧģ�Ͳ��õ����ַ���ģ�͡����ģ�Ϳ��Լ�Ϊÿ�����̾�Ϊʽ(1)��ʾ��ʽ�ķ�������ɵķ����飺
![]() (1)
(1)
���У�xi(t)Ϊ����i��ʱ��t��Ũ�ȣ�NΪ���������������������wijΪ����j�Ի���i����Ӱ���Ȩֵ���������ֵ������ʱ��ʾ�����أ�����ʱ��ʾ�����أ�0��ʾ��һ����ֵ��Χ�ڿ�����Ϊ����������ã�bi��ʾ��û�е������������»���i�ı仯��Ҳ���������Ի���i�Ĵ̼��������ı���Ũ�ȵı仯��
��ʵ���У������ṩһЩ�㶨�����̼�����bi�Ǻ㶨�ġ�Ϊ�˷��㣬�����ʱ���趨biΪ0������ʩ���κ����̼���Ȼ��N��������T��ʱ��ν��в��������õ��������ݣ�������֯��ʽ���£�
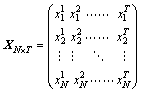 (2)
(2)
���У�xΪ�������Ũ�ȣ��±��ʾ�����ʶ���ϱ��ʾ����ʱ�����У�����N������T��ʱ����Ե㡣����ʽ(2)��ʽ(1)����д��ʽ(3)��ʾ��ʽ��
![]() (3)
(3)
Ϊ�����Ȩֵ����W�������ѷ�������б��κĽ����������һ���̶�����߾��ȣ���Ҫ�Ӹ�������߾�����Ҫ��������ֵ�ֽ⡣���Խ�X�����������ֵ�ֽ⣺
![]() (4)
(4)
���У�����A��һ�������������������ĶԽǾ���Ϊ�˼��㷽�㣬�����ڼ�������н��е�����ʹ�þ���A�жԽ����ϵ�ֵ�ǰ��մ�С��������ġ�������ֵ��1��ֵ�������ֵ�ݼ�������ļ����Խ����ϵ�ֵΪ0���ǶԽ����ϵ�ֵȫΪ0���Խ�������ֵ�ĸ�������Ϊ0�����˾������Ϊ���Ⱦ�����ʽ(4)�У�����ֵ�ֽ�õ��Ľ��U��V�������������Ҷ����㣺
![]() (5)
(5)
ʽ�У�EΪ��λ����
��ʽ(3)����ͬʱ��BN��T��Ȼ��������ͬʱ�ҳ�XN��T���������ʽ(4)����ʽ(3)���õ���
![]() (6)
(6)
��ʽ(6)�У�������ÿ��������������ر��Ǿ���U������������ָ�ľ��ǹ��������[7]���ڹ�����������£�����һ��������������������������������Ƿ�������������
��ʽ(6)�õ�����������繹�������Ȩֵ����W���о�����һ��ͰѴ��ؽ���Ϊ���յ������������ý��������ȷ����Ϊ����ʽ(6)������ֵ�ܶ࣬�������ܴ����������⣬��N>>T�����������ֵ��ʵ����һ�������Ž⡣�ڼ�������У�����ܹ������ܶ�ĵõ��⼯����ȡֵ��Χ����ʹ���Ż��㷨���м��㡣�����㷨���ؽ�Ļ����ϣ�������ʽ����ͨ�⣺
![]() (7)
(7)
ʽ�У�CΪ���ⳣ����
���������ͨ��Ϳ��Եõ�������ȡֵ��Χ���Ӷ��������ַ���ģ�ͣ��������Ż����ڴ˷�Χ������������硣
��ʽ(1)������Ȩֵ����W���б��Ρ�������Ҫ������㷨�������������������С�������������Ϊ����ֵ�۲�ֵ֮��IJ
![]() (8)
(8)
���У�rikΪʽ(1)�IJл���vikΪrik��1��Ȩֵ����
![]() (9)
(9)
��ʽ(9)���Կ�����rik���ϲл����������Ҫ���ľ�����ʽ(7)��ʾ������Ȩֵȡֵ��Χ��ʹ�л���С�����õ���Ҫ���е�Ŀ��֮����Ҫ�ѷ��̽�һ���Ľ����Ա����Ч�ʣ���������ʽ(8)��(9)�У���������ֻ��wij���������й���N��������Ȩֵ����WΪһ�������������IJ���ΪN����һ�������������Ӵ����������IJ�����������ָ�������������ʺ���⣬��ˣ���Ҫ��Ȩֵ����W��һ���Ż����ֽ⣬����Ȩֵw����Ϊ��
![]() (10)
(10)
����ʽ(10)��ת��֮��Ȩֵ����W�е�ֵֻ��t��x��2��������أ��Ӷ�ʹ�ü���dz���㡣
�Ż��㷨���棬���IJ�������Ⱥ�㷨[8]���Ŵ��㷨[9]���ϵķ�ʽ��ͨ����С��ʽ(8)�������Сֵ��Ȼ����֤�Ƿ����ʽ(7)�ķ�ʽ��������յ����硣
2 �㷨��ʵ��
�����㷨��ʵ�ֲ�����Java���Ա�д�����û���ΪMyEclipse 8.0�汾���㷨�����̲������¡�
(1) ��ȡ����������ݣ����ʽ���Ϲ��ʱ������ݱ�(MIAME[10])��
(2) ���Ѿ���ȡ�Ļ���������������ɵľ�������ʽ(4)��������ֵ�ֽⲢ�õ�ʽ(4)�е�U��V��
(3) ����ʽ(6)�����������Ȩֵ����W���ؽ�W����������ʽ(7)���W��ͨ��Wͨ��
(4) ������ɴ���Ȩֵ��������Ⱥ�㷨���Ŵ��㷨�ij�ʼȺ�塣�����ݲ���(3)�õ���ȡֵ��Χ��̭������Ҫ������Ȩֵ����̭��Ȩֵ�������ɣ�ֱ�����е����Ȩֵ����Wͨ������Ϊֹ��
(5) ����ʽ(10)�ֽ�Ȩֵ����Ȩֵ��άΪֻ��2�������ķ����顣�������Ŵ��㷨������Ⱥ�㷨(PSO)��С��ʽ(8)���õ���Сֵ��
(6) ������(5)����õ���ֵ����Wͨ���������㷨��ֹ��������벽��(5)��
�㷨����ͼ��ͼ1��ʾ��
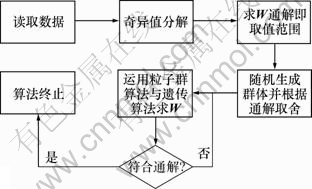
ͼ1 �����㷨����ͼ
Fig.1 Flow chart of proposed algorithm
3 ʵ���������
3.1 ���Է���
Ϊ�˲��Ա����㷨��Ч�������бȽ�ʵ�顣�Ƚ϶���Ϊ����ʹ��ʽ(1)�Ļ����ַ���ģ��(����1)�ʹ���ʽ(10)�ĸĽ����ַ���ģ��(����2)������ģ��(����3)����3��ģ�͵ıȽ�ʵ�齫��Ϊģ������ʵ�����ʵ����ʵ��2���֡�
����Bansal��[11]�����������Ԥ��ֵ(PPV)�����ж�(SE)�����㷨���ӡ�
3.2 ģ������ʵ��
������Դ���������ޱ������[12]���ַ��������ƻ���������ݵķ������м����á���������ɵ�������ȣ��ޱ����������ϻ���������������ѧ���������ɵ��ޱ�������У�ÿ���ڵ�����������ӽ������ʷֲ�p(k)~k�������������ص��������㷨[13]���й���������r�趨Ϊ3���ڵ���NΪ20�����������㷨�Ͳ���������������ͼ2��ʾ����ͼ2�У�ԭ��Ϊ�ڵ�0����ʱ������Ϊ�ڵ�1��2������20�������Ϊ�����߶�ĩ�ˣ�ָ���ؽڵ㡣���������Ժ���ʽ(1)�����������ƶϣ����е�bi������Ϊ0�����ݵ�ʱ����趨Ϊ50��������������趨Ϊ-0.5~0.5֮�䡣
��ͼ2��ʾ��ͼģ�������������ݺ�������õ������ͼ3��ʾ����ͼ3�У�����ʾ�ĺ�����ͼ2��һ������ͬ����ԲȦ�����������ҵ��ع�ϵ��
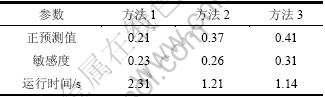
Ϊ�˱������ɺϣ����г���100�Σ�����N��ȡֵ��Ϊ20�������ȡƽ��ֵ�������㷨�����ӡ�3�ַ����ıȽϽ�����1��ʾ��
�ӱ�1��֪�������㷨(������3)����Ԥ��ֵ�����ж����Ը�������2���㷨�Ľ����������ʱ��Ҳ������2�ַ���������ʱ��̡�
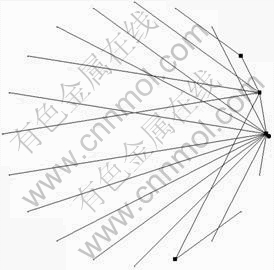
ͼ2 ����r=3��N=20���ޱ������ͼ
Fig.2 Scale-free network with parameter r=3, N=20

ͼ3 ����ͼ2�����ɵ�����������Ļ����������ͼ
Fig.3 Gene regulatory network calculated with proposed algorithm from data simulated by Fig. 2
��1 3���㷨ģ������Ч���Ƚ�
Table 1 Compared results of three kinds of algorithm in simulation data
����2�ȷ���1������ʱ���ϸ������Ƶ�ԭ�����ڷ���2�ѷ���1�е�N2����������Ϊֻ��2��������ʹ������ʱ�������̡����ķ���Ϊ����2�ṩ��һ���ܺõĺ�ѡ��ļ��ϣ������˷���2Ѱ�ŵĿռ������ʱ�䣬���ǣ���������ֵ�ֽ����㱾��������һ����ʱ�䣬��ˣ��뷽��2������ʱ����ȣ����ķ���������Ч��������ߡ�
3.3 ��ʵ����ʵ��
��ʵ���ݵļ�����õ���GEO[14]����������ݿ������صĻ���������ݣ�������õ��Ǿ���Ľ�ĸ����������ݣ�������GDS38���ݵ��Ӽ��������Ľ��ͼ���Դ��ձ�KEGG[15]���ݿ��е�pathway��õ���������ݰ���3���Ӽ�����3���Ӽ�����3��ʵ��������Alpha��elu��cdc15�����Ƿֱ����18��14��24��ʱ��㡣��ȡ������1�������Ӽ���ͼ4��ʾ�����������㷨���㣬��Ӧͼ4�����ɵĻ������������ͼ5��ʾ����ͼ5�У����еĺ�����ͼ2�е�һ����ԭ����Ϊ��1����ʼ�㣬ԲȦ��ʾ�Ե��أ������ʾ�����ضΡ�
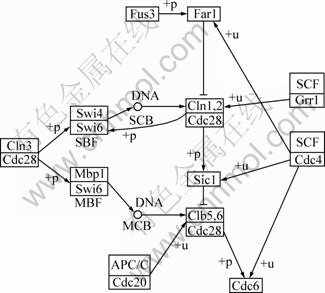
ͼ4 KEGG���ݿ��pathway�н�ĸ������������1���Ӽ�ͼ
Fig.4 A part of yeast cell cycle pathway available from KEGG
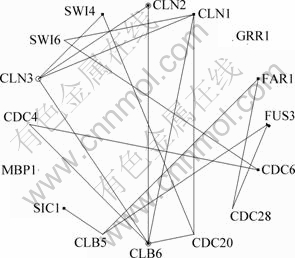
ͼ5 �ñ����㷨��õĻ����������ͼ(��Ӧͼ4)
Fig.5 Gene regulated network calculated by proposed algorithm (according to Fig. 4)
����ʽ(1)��(10)���ַ���ģ�ͣ�����100�κ�ȡƽ��ֵ�����ս�����2��ʾ��
�ӱ�2���Կ����������㷨(������3)������2���㷨����Խ��������ʱ���������������㷨������2���㷨��ȸ�С����ģ��ʵ������ȣ����Ʋ��Ǻ����ԡ�
��2 3���㷨������ʵ���ݽ���Ƚ�
Table 2 Compared results of three kinds of algorithm in real data

4 ����
(1) �����һ�־���ͨ��Ĺ���������������㷨�������µ��ں��㷨����������������ṩ��һ����ѡ��ļ��ϣ�ʹ�������������ȥ�˷ǽ�IJ���Ҫ���㣬����������ʱ�䣬ͬʱ��ʹ������������ȷ��ͨ��������2�ִ�ͳ�ַ���ģ���㷨�ĶԱ�ʵ�飬֤ʵ�˱����㷨��Ч�ʽϸߣ�����Ԥ�ڽ����
(2) �����㷨��Ŀ���Ǵ����������еõ���ӽ�����ʵ�Ļ����������Ľ�������ʵ��ˮƽ�����ǣ����ĵĻ�����������㷨����ľ��ȣ������һ����ߣ����д���һ���о���
�ο����ף�
[1] D��haeseleer P, Liang S, Somogyi R. Genetic network inference: from co-expression clustering to reverse engineering[J]. Bioinformatics 2000, 16: 707-726.
[2] Dojer N, Gambin A, Mizera A, et al. Applying dynamic Bayesian networks to perturbed gene expression data[J]. BMC Bioinformatics, 2006, 7: 249.
[3] Beal M J, Falciani F, Ghahramani Z, et al. A Bayesian approach to reconstructing genetic regulatory networks with hidden factors[J]. Bioinformatics, 2005, 21: 349-356.
[4] Bansal M, Gatta G D, di Bernardo D. Inference of gene regulatory networks and compound mode of action from time course gene expression profiles[J]. Bioinformatics, 2006, 22: 815-822.
[5] Eugene N, Emmanuel B. Regulatory network reconstruction using an integral additive model with flexible kernel functions[J]. BMC Systems Biology, 2008, 2: 8.
[6] Nelson P A, Kahana Y. Spherical harmonics, singular-value decomposition and the head-related transfer function[J]. Journal of Sound and Vibration, 2001, 239(4): 607-638.
[7] Liang M L, Dai L F. The left and right inverse eigenvalue problems of generalized reflexive and anti-reflexive matrices[J]. Journal of Computational and Applied Mathematics, 2010, 234: 743-749.
[8] Brits R, Engelbrecht A P, van den Bergh F. Locating multiple optima using particle swarm optimization[J]. Appl Math Comput, 2007, 189(2): 1859-1883.
[9] Chen D Y, Chuang T R, Tsai S C. Jgap: A java-based graph algorithms platform[J]. Software Pract Exper, 2001, 31(7): 615-635.
[10] Tim F R, Philippe R S, Paul T S, et al. A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB[J]. BMC Bioinformatics, 2006, 7: 489.
[11] Bansal M, Belcastro V, Ambesi-Impiombato A, et al. How to infer gene networks from expression profiles[J]. Mol Sys Biol, 2007, 3: 78.
[12] Barab��si A L, Albert R. Emergence of scaling in random networks[J]. Science, 1999, 286: 509-512.
[13] Silva A C, da Silva J K L, Mendes J F F. Scale-free network with Boolean dynamics as a function of connectivity[J]. Phys Rev E, 2004, 70(6): 66140-66147.
[14] Wilhite S E, Barrett T. Strategies to explore functional genomics data sets in IVCBI��s GEO database[J]. Methods Mol Biol, 2012, 802: 41-53.
[15] KEGG Cell cycle-yeast-Saccharomyces cerevisiae[EB/OL]. [2010-04-13].http://www.genome.jp/kegg/pathway/sce/sce04111.html.
(�༭ �Կ�)
�ո����ڣ�2011-05-10�������ڣ�2011-07-23
������Ŀ��������Ȼ��ѧ����������Ŀ(60873146��60973092��60903097)�����Ҹ����о���չ�ƻ�(��863���ƻ�)��Ŀ(2009AA02Z307)�����ִ�ѧ�о������»���������Ŀ(20111062��200903175)������ʡ�Ƽ�����Ŀ(20090116��20101589)
ͨ�����ߣ�����(1979-)���У����ֳ����ˣ���ʿ����ʦ�����¼������ܺ�������Ϣѧ�о����绰��0431-88499590��E-mail��zyou@jlu.edu.cn
ժҪ���ڸĽ��ֽ�Ȩֵ������ַ���ģ�ͻ����ϣ���������ֵ�ֽⷽ�����������㡣���㷨����ͨ������ֵ�ֽ���ؽ�õ���������ս��������ͨ������õ�ͨ���ṩ��ѡ�⼯�ķ�����Ϊ�ַ���ģ���㷨��������ʱ�����߽�����ȡ��������㷨��������ͳ�ַ���ģ���㷨���жԱȣ���֤������������㷨��Ч�ʽϸߡ�
