

![]()

Trans. Nonferrous Met. Soc. China 22(2012) 654-660
First-principle calculations of ductile CeAg intermetallic compound
SHI Yao-jun1, 2, DU Yu-lei1, 2, CHEN Guang1, 2
1. Engineering Research Center of Materials Behavior and Design of Ministry of Education,
School of Materials Science and Engineering, Nanjing University of Science and Technology, Nanjing 210094, China;
2. Jiangsu Institute of Advanced Materials, Danyang 212300, China
Received 9 September 2011; accepted 12 January 2012
Abstract:
The first-principle calculations were performed to investigate the structural, mechanical, electronic and thermal properties of the binary ductile intermetallic compound CeAg with B2 (CsCl) structure. The calculated value of lattice constant a0 for CeAg with generalized gradient approximation is 3.713 ?, which is in better agreement with experimental data than local spin density approximation. The negative energy of formation implies that CeAg with B2 structure is thermodynamically stable phase. The greater separation between the d bands of Ce and Ag results in weaker bond hybridization of Ce d��Ag d, which prevents formation of directional covalent bonding. The three independent elastic constants (C11, C12 and C44) are derived and the bulk modulus, shear modulus, elastic modulus, anisotropy factor, and Poisson ratio are determined to be 57.6 GPa, 15.8 GPa, 43.4 GPa, 3.15 and 0.374, respectively. The elastic constants meet all the mechanical stability criteria. The value of Pugh��s criterion is 3.65. The ductility of CeAg is predicted if Pugh��s criterion is greater than 1.75. Furthermore, the variations of volume, bulk modulus, heat capacity, and thermal expansion coefficient with temperature and/or pressure were calculated and discussed.
Key words:
CeAg; first-principle; elastic constant; thermodynamic property;
1 Introduction
Many binary intermetallic compounds (Ni3Al, NiAl and FeAl, etc) have a combination of low density, good oxidation resistance and high strength at elevated temperatures. These excellent properties make them potentially useful for high-temperature structural materials, but binary polycrystalline intermetallic compounds usually possess little or no ductility at ambient temperatures if they are fully ordered with stoichiometric compositions [1]. Recently, a new family of ductile intermetallic compounds has been discovered. They are fully ordered, stoichiometric binary rare-earth B2-type (CsCl-type structure) intermetallic compounds with formula RM, where R is a rare-earth element and M is a main group or late transition metal [2]. Then, many experimental works have been done to reveal the mechanism of high ductility for RM [3-5]. Their results indicate that multiple slip systems, fine grain size, and rare-earth element are factors which may contribute to high ductility of RM. However, up to now, the mechanism has not been understood completely. LnAg (Ln=rare earth element) is a typical family of ductile RM. Cerium is the first element with 4f electron of Ln group elements, and CeAg is an important member of the ductile RM intermetallic compounds. The magnetic properties of CeAg have been investigated widely both experimentally [6-8] and theoretically [9, 10] due to its unusual magnetic properties at low temperature and high pressure. However, the electronic structure, thermodynamic properties and ductile mechanism have not been understood completely. To get a better understanding of the anomalous ductility of CeAg, more fundamental studies of its electronic structural properties are required.
In the present work, the first principle calculations are performed to investigate the fundamental properties of the RM compound CeAg with B2 structure including total energy, lattice constant, formation energy, electronic band structure, density of states, mechanical properties and thermodynamic properties. The phase stability, bonding character and account for high ductility were discussed on the basis of the calculation results.
2 Computational methods
The first-principle calculations were based on the density functional theory (DFT). The total energies were calculated using full-potential linearized augmented plane-wave method (FP-LAPW) plus local orbital program as implemented in the WIEN2k package [11], and with the generalized gradient approximation (GGA) of PERDEW, BURKE and ERNZRHOF (PBE) [12] for the exchange-correlation energy. The spherical harmonics inside the muffin-tin are taken with an angular momentum lmax=10 while the charge density is Fourier expanded up to Gmax=12. The muffin-tin sphere radii (Ri) of Ce and Ag was chosen as 2.7 and 2.5 a.u., respectively. The basis function was expanded up to RminKmax=9.0 in order to achieve convergence, where Rmin is the minimum sphere radius and Kmax is the maximal value of the reciprocal lattice vector used in the plane wave expansion. Integration in the reciprocal space was performed by using the tetrahedron method with a k mesh of 12��12��12 k-points in the irreducible wedge of the Brillouin zone (BZ). The total energy is converged to within 0.01 mRy/unit cell during the self-consistency cycle.
The structure of CeAg was firstly optimized and then the elastic constants were calculated by the strain��energy method. An set of strains were applied to the unit cell lattice of optimized structure. Then, the elastic constants were determined from the resulting change in total energy on the deformation. For the cubic crystals, there are three independent elastic constants, which are usually referred to as C11, C12 and C44. Three strains were employed to determine all the elastic constants. The detailed description of this method can be found in Ref. [13].
To investigate the thermodynamic properties of CeAg, the quasi-harmonic Debye model was applied [14]. The non-equilibrium Gibbs energy G*(V; P, T) can be written as:
![]() (1)
(1)
where E(V) is the total energy per unit cell; ��D(V) is the Debye temperature; Avib is the vibrational Helmholtz free energy, and can be written using the Debye model of the phonon density of states as [15-19]:
![]() (2)
(2)
where D(��D/T) represents the Debye integral; ��D is the Debye temperature; n is the number of atoms per formula unit. Assuming an isotropic solid, ��D can be computed as [15]:
![]() (3)
(3)
where Mr is the relative molecular mass per formula unit; f(��) was given in Refs. [17,18]; BS is the adiabatic bulk modulus, which is approximately given by the static compressibility.
![]() (4)
(4)
Thus, the non-equilibrium Gibbs function G*(V; P, T) can be minimized with respect to volume V:
![]() (5)
(5)
By solving Eq. (5), the thermal equation of state (EOS) V(P,T) can be obtained. The specific heat capacity cV and the thermal expansion coefficient �� are given by
![]() (6)
(6)
![]() (7)
(7)
where �� is the Gr��neisen parameter defined as
![]() (8)
(8)
Through the quasi-harmonic Debye model, thermodynamic parameters including the bulk modulus, thermal expansion coefficient and specific heats at constant volume, heat capacity cV under different temperatures and/or pressures can be obtained from the calculated E��V data. A detailed description of this method can be found in Ref. [14].
3 Results and discussion
3.1 Crystal structure parameters
CeAg with B2 CsCl-type has two atoms in one unit cell with ![]() space group. Figure 1(a) illustrates the crystal structure of CeAg. The Ce atoms are positioned at (0, 0, 0), Ag atoms at the centre of unit cell (0.5, 0.5, 0.5) [20]. Before starting the calculations of electronic structure and the lattice parameters are optimized with different approximations to the exchange-correlation energy for a comparison. Figures 1(b) and (c) show the variation of total energy with volume for the local spin density approximation (LSDA) [21] and GGA, respectively. The calculated values of the crystal lattice constants and equilibrium unit cell volume, as well as the corresponding experimental results for CeAg are given in Table 1. The computed lattice parameter values are 3.595 ? with LSDA and 3.713 ? with GGA, respectively. Our theoretical lattice constant a0 for CeAg with GGA is only 0.88% smaller than the experimental one, while it is 4.03% with LSDA. Hence, the theoretical lattice constant with GGA is used in all calculations.
space group. Figure 1(a) illustrates the crystal structure of CeAg. The Ce atoms are positioned at (0, 0, 0), Ag atoms at the centre of unit cell (0.5, 0.5, 0.5) [20]. Before starting the calculations of electronic structure and the lattice parameters are optimized with different approximations to the exchange-correlation energy for a comparison. Figures 1(b) and (c) show the variation of total energy with volume for the local spin density approximation (LSDA) [21] and GGA, respectively. The calculated values of the crystal lattice constants and equilibrium unit cell volume, as well as the corresponding experimental results for CeAg are given in Table 1. The computed lattice parameter values are 3.595 ? with LSDA and 3.713 ? with GGA, respectively. Our theoretical lattice constant a0 for CeAg with GGA is only 0.88% smaller than the experimental one, while it is 4.03% with LSDA. Hence, the theoretical lattice constant with GGA is used in all calculations.

Fig. 1 Crystal structure of CeAg (a), variation of total energy with volume for LSDA approximation (b) and variation of total energy with volume for GGA approximation (c)
Table 1 Theoretical and experimental lattice constants as well as unit cell volumes of CeAg in B2 cubic phase

3.2 Energy of formation
The energy of formation is known to be a measure of the phase thermodynamic stability, so the energy of formation (��Eform) for CeAg is calculated according to the following expression [22]:
��Eform=![]() [Etot(CeAg)-Etot(Ce) -Etot(Ag)] (9)
[Etot(CeAg)-Etot(Ce) -Etot(Ag)] (9)
where Etot(CeAg), Etot(Ce) and Etot(Ag) are the total energies for CeAg (space group ![]() , 2 atom/cell, Ce (space group
, 2 atom/cell, Ce (space group ![]() , 1 atom/cell) and Ag (space group
, 1 atom/cell) and Ag (space group ![]() , 1 atom/cell), respectively. The calculated value for CeAg is -0.04 eV/atom. The negative energy of formation implies that CeAg with B2 structure is thermodynamically stable phase.
, 1 atom/cell), respectively. The calculated value for CeAg is -0.04 eV/atom. The negative energy of formation implies that CeAg with B2 structure is thermodynamically stable phase.
3.3 Electronic properties
Figure 2 exhibits the calculated energy band structure of CeAg at equilibrium lattice parameters along selected high-symmetry directions within the first Brillouin zone. The valence bands of CeAg can be divided into two groups, i.e., a low-energy group mainly composed of Ag 4d states, and the upper valence bands composed of Ce 4f and 5d states. As shown in Fig. 2, valence and conduction bands overlap considerably at the Fermi level and as a result there is no energy gap at EF. This suggests that CeAg exhibits a nearly metallic character.
The total and partial densities of states (DOS) for CeAg are calculated using the optimized structure at the equilibrium volume. As shown in Fig. 3, the lowest-lying states from -6 to -4 eV are derived from Ag 4d states. The states above the Fermi level are mainly due to Ce 5d and 4f states. The d bands of Ce are diffuse, while the d bands of Ag are much narrower than those in pure FCC Ag crystal. The greater separation between the d bands of Ce and Ag results in weaker bond hybridization of Ce d��Ag d, which prevents formation of directional bonding. This suggests that CeAg would exhibit high ductility like some other RM intermetallics [23].
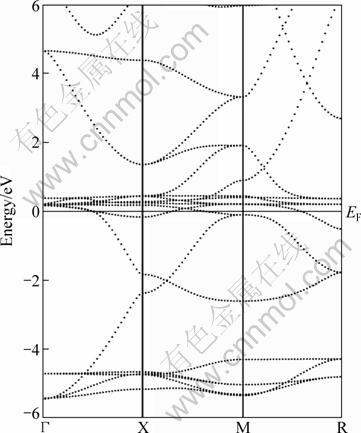
Fig. 2 Electronic band structure of CeAg
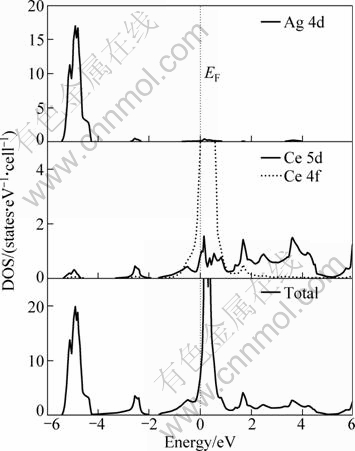
Fig. 3 Total and partial densities of states for CeAg
3.4 Mechanical properties
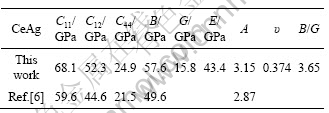
The elastic constants are essential for understanding macroscopic mechanical properties of crystal as they relate to various fundamental solid state properties and thermodynamic properties. Table 2 lists the values of the single crystal elastic constants, the bulk modulus B, the shear modulus G, elastic modulus E, anisotropy factor A and Poisson ratio ��. Furthermore, the value of Pugh��s criterion is also listed. The bulk modulus B and Voigt-Reuss-Hill averaged shear modulus GH [24] were obtained through the following equations: B=(C11+2C12)/3 and GH=(GV+GR)/2, where the effective Voigt shear modulus GV and Reuss shear modulus GR are
![]()
![]() (10)
(10)
The elastic modulus E, Poisson ratio �� and anisotropic factor A are given as:
![]()
![]()
![]() (11)
(11)
As shown in Table 2, the calculated elastic constants and bulk modulus are close to values reported by MORIN [6]. The elastic stability is a necessary condition for a crystal to exist. The requirements of mechanical stability for a cubic crystal lead to the following restrictions on the elastic constants [25]: C11>0, C44>0, C11-C12>0, C11+2C12>0, C11>B>C12. Obviously, the calculated elastic constants satisfy the mechanical stability criterion, which means that CeAg is stable in B2 structure, and in good agreement with the analysis of formation energy. PUGH [26] proposed that materials with melting temperatures above 900 ��C is predicted to be in ductile behavior if the value of Pugh��s criterion B/G>1.75 [27]. Obviously, our calculated value satisfies criterion. The ductility of CeAg is predicted, which is consistent with the observed ductility in experiments [28].
Table 2 Calculated elastic constants, bulk modulus, shear modulus, elastic modulus, anisotropy factor, Poisson ratio of CeAg
3.5 Thermodynamic properties
Figure 4 shows the normalized volume��pressure diagram at 300, 600 and 1000 K for CeAg. The unit cell volume decreases smoothly at different temperatures; no abrupt changes occur with increasing pressure. This change of curves can be attributed to interaction between atoms and thermal expansion. Furthermore, the normalized volume decreases faster with increasing temperature.

Fig. 4 Normalized volume��pressure diagram for CeAg at different temperatures
The temperature dependence of bulk modulus at 0 GPa of pressure for CeAg is shown in Fig. 5(a). From 0 K to 600 K, bulk modulus decreases linearly with increasing temperature. The bulk modulus of CeAg drops by 18% from 0 K to 1000 K. The relationship between bulk modulus and pressure for CeAg at different temperatures is shown in Fig. 5(b). From 0 to 18 GPa, the bulk modulus increases linearly with increasing pressure. The B values of CeAg at 300, 600 and 1000 K increase by 148%, 163% and 165%, respectively. Otherwise, the bulk modulus decreases with increasing the temperature at a fixed pressure.

Fig. 5 Bulk modulus as function of temperature at p=0 GPa (a) and pressure dependency of bulk modulus for CeAg at 300, 600 and 1000 K (b)
The calculated specific heat capacity at constant volume cV, versus temperature for CeAg at a pressure of 0 GPa is shown in Fig. 6(a). The cV increases rapidly in the low temperature region below 200 K and almost approaches a constant above 400 K due to the anharmonic approximation of the Debye model in the high temperature region. The cV obeys the expected T3 power law in the low temperature limit, and it is very close to the Dulong-Petit limit of cV=3nNkB=49.884 J/(mol��K). As is known to all, 3nNkB is commonly satisfied with all solids at higher temperatures. Figure 6(b) illustrates the variation of the volumetric thermal expansion coefficient ���� of CeAg as a function of the temperature at a pressure of 0 GPa. It shows that initially, the ���� increases rapidly with increasing temperature before leveling off at T>200 K. The change of ���� is very small at high temperature, which is similar to specific heat capacity at constant volume cV.
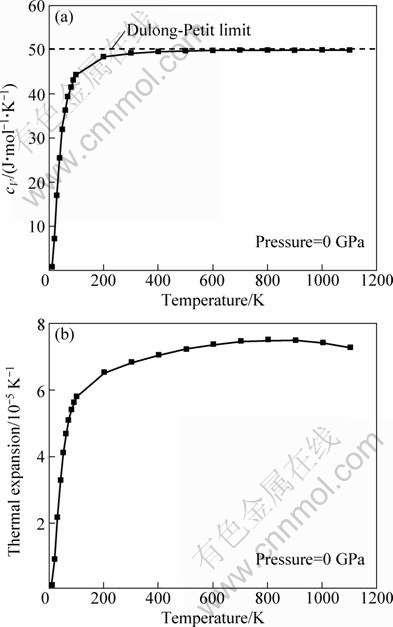
Fig. 6 Calculated heat capacity values at constant volume cV versus temperature (a) and thermal expansion versus temperature (b) for CeAg at P=0 GPa
4 Conclusions
1) The calculated lattice parameters using GGA method are in better agreement with the experimental data than LSDA method for CeAg.
2) The calculated formation energy for CeAg is -0.04 eV/atom. The negative energy of formation indicates that CeAg with B2 structure is thermodynamically stable phase.
3) Band structure and density of states show that the greater separation between d bands of Ce and Ag results in weaker bond hybridization of Ce d��Ag d. This would prevent formation of directional covalent bonding and explain high ductility of CeAg.
4) Calculated elastic constants satisfy the mechanical stability criterion and the ductility of CeAg is predicted by Pugh��s criterion.
5) The thermodynamic properties are analyzed using the quasi-harmonic Debye model. The results show that volume and bulk modulus vary linearly with increasing temperature. Furthermore, specific heat capacity cV is close to the Dulong-Petit limit, which is common to all solids at high temperatures.
References
[1] RUSSELL A M. Ductility in intermetallic compounds [J]. Advanced Engineering Materials, 2003, 5: 629-639.
[2] GSCHNEIDNER K, RUSSELL A, PECHARSKY A, MORRIS J, ZHANG Z H, LOGRASSO T, HSU D, LO C H C, YE Y Y, SLAGER A, KESSE D. A family of ductile intermetallic compounds [J]. Nature Materials, 2003, 2: 587-590.
[3] ZHANG Z, RUSSELL A M, BINER S B, GSCHNEIDNER K A Jr, LO C C H. Fracture toughness of polycrystalline YCu, DyCu, and YAg [J]. Intermetallics, 2005, 13: 559-564.
[4] RUSSELL A M, ZHANG Z, LOGRASSO T A, LO C C H, PECHARSKY A O, MORRIS J R, YE Y, GSCHNEIDNER K A, SLAGER A J. Mechanical properties of single crystal YAg [J]. Acta Materialia, 2004, 52: 4033-4040.
[5] CAO G H, SHECHTMAN D, WU D M, BECKER A T, CHUMBLEY L S, LOGRASSO T A, RUSSELL A M, GSCHNEIDNER K A Jr. Determination of slip systems and their relation to the high ductility and fracture toughness of the B2 DyCu intermetallic compound [J]. Acta Materialia, 2007, 55: 3765-3770.
[6] MORIN P. Quadrupolar ordering in CeAg [J]. Journal of Magnetism and Magnetic Materials, 1988, 71: 151-164.
[7] TAKKE R, DOLEZAL N, ASSMUS W, L?THI B. Magnetic and elastic properties of CeAg [J]. Journal of Magnetism and Magnetic Materials, 1981, 23: 247-253.
[8] MONACHESI P, MORONI E G. Pressure induced magnetic moment reduction in CeAg [J]. Solid State Communications, 1990, 74: 1349-1353.
[9] FRAIZZOLI S, MONACHESI P, MORONI E G. Localized behavior of f electrons in CeAg: A theoretical study [J]. Journal of Applied Physics, 1991, 69: 4659-4661.
[10] MONACHESI P, ANDREANI L C, CONTINENZA A, MCMAHAN A K. Volume dependence of Anderson hybridization in cubic CeCd and CeAg [J]. Journal of Applied Physics, 1993, 73: 6634-6636.
[11] BLAHA P, SCHWARZ K, MADSEN G K H, KVASNICKA D, LUITZ J. WIEN2k, An augmented plane wave plus local orbitals program for calculating crystal properties [M]. Vienna: Techn Universit?t Wien, 2011.
[12] PERDEW J P, BURKE K, ERNZERHOP M. Generalized gradient approximation made simple [J]. Physical Review Letters, 1996, 77: 3865-3868.
[13] CHARPIN T. A package for calculating elastic tensor of a cubic phases using WIEN [M]. Paris: Laboratoire Des G��omat��riaux de l��IPGP, 2001.
[14] BLANCO M A, FRANCISCO E, LUANA V. Gibbs: isothermal-isobaric thermodynamics of solids from energy curves using a quasi-harmonic Debye model [J]. Computer Physics Communications, 2004, 158: 57-72.
[15] BLANCO M A. M��todos cu��nticos locales para la simulaci��n de materiales i��nicos. Fundamentos, algoritmos y aplicaciones [D]. Spain: Universidad de Oviedo, 1997.
[16] BLANCO M A, PEND?S A MART?N, FRANCISCO E, RECIO J M, FRANCO R. Thermodynamical properties of solids from microscopic theory: Applications to MgF2 and Al2O3 [J]. Journal of Molecular Structure: Theochem, 1996, 368: 245-255.
[17] FRANCISCO E, RECIO J M, BLANCO M A, PEND?S A M. Quantum-mechanical study of thermodynamic and bonding properties of MgF2 [J]. The Journal of Physical Chemistry A, 1998, 102: 1595-1601.
[18] FRANCISCO E, SANJURJO G, BLANCO M A. Atomistic simulation of SrF2 polymorphs [J]. Physical Review B, 2001, 63: 0941071-0941079.
[19] FL?REZ M, RECIO J M, FRANCISCO E, BLANCO M A, PEND?S A M. First-principles study of the rocksalt-cesium chloride relative phase stability in alkali halides [J]. Physical Review B, 2002, 66: 1141121-1141128.
[20] VILLARS P, CALVERT L D. Pearson��s handbook of crystallographic data for intermetallic phases, vols. 1�C4 [M]. Materials Park: ASM International, 1991.
[21] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects [J]. Physical Review A, 1965, 140: 1133-1138.
[22] SHEIN I R, SHEIN K I, IVANOVSKII A L. First-principles study on the structural, cohesive and electronic properties of rhombohedral Mo2B5 as compared with hexagonal MoB2 [J]. Physica B, 2007, 387: 184-189.
[23] GSCHNEIDNER K A Jr, JI M, WANG C Z, HO K M, RUSSELL A M, MUDRYK Y A, BECKER A T, LARSON J L. Influence of the electronic structure on the ductile behavior of B2 CsCl-type AB intermetallics [J]. Acta Materialia, 2009, 57: 5876-5881.
[24] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proceedings of the Physical Society. Section A, 1952, 65: 349-354.
[25] BECKSTEIN O, KLEPEIS J E, HART G L W, PANKRATOV O. First-principles elastic constants and electronic structure of ��-Pt2Si and PtSi [J]. Physical Review B, 2001, 63: 1341121-1-12.
[26] PUGH S F. XCII. Relations between the elastic moduli and the plastic properties of polycrystalline pure metals [J]. Philosophical Magazine, 1954, 45: 823-843.
[27] MORRIS J R, YE Y Y, LEE Y B, HARMON B N, GSCHNEIDNER K A, RUSSELL A M. Ab initio calculation of bulk and defect properties of ductile rare-earth intermetallic compounds [J]. Acta Materialia, 2004, 52: 4849-4857.
[28] WOLLMERSHAUSER J A, KABRA S, AGNEW S R. In situ neutron diffraction study of the plastic deformation mechanisms of B2 ordered intermetallic alloys: NiAl, CuZn and CeAg [J]. Acta Materialia, 2009, 57: 213-223.
���Խ����仯����CeAg�ĵ�һ��ԭ������
ʷҫ��1, 2��������1, 2���� ��1, 2
1. �Ͼ�������ѧ ���Ͽ�ѧ�빤��ѧԺ��������������ƽ����������о����ģ��Ͼ� 210094��
2. ����ʡ(����)�����ܺϽ�����о�Ժ������ 212300
ժ Ҫ�����õ�һ��ԭ�����㷨�о�����B2 (CsCl) �ṹ�Ķ�Ԫ���Խ����仯����CeAg�Ľṹ����ѧ���ܡ����������Լ�����ѧ���ܡ����ù����ݶȽ��Ƽ���õ��ľ�����Ϊ3.713 ?���������ܶȽ��Ƽ���Ľ��������ʵ��ֵ�������γ��ܱ�������B2�ṹ��CeAg������ѧ�ȶ��ࡣCe��Agԭ�ӵ�d�ܴ�����뵼�����ߵļ����ӻ����ý������Ӷ���ֹ���з����ԵĹ��ۼ��γɡ�����õ���CeAg��3���������Գ���(C11��C12��C44)����ģ��������ģ��������ģ�����������������Լ����ɱȷֱ�Ϊ57.6 GPa�� 15.8 GPa�� 43.4 GPa��3.15��0.374�����Գ�������������ѧ�ȶ�����CeAg��Pugh�о�ֵΪ3.65����Pugh�о�ֵ����1.75ʱ��CeAg�������õ����ԡ����⣬�������������CeAg���������ģ�������ݺ�������ϵ�����¶Ȼ���ѹ���ı仯���ɡ�
�ؼ��ʣ�CeAg����һ��ԭ�������Գ���������ѧ����
(Edited by LI Xiang-qun)
Foundation item: Project (2011CB605504) supported by the National Basic Research Program of China; Project (50871054) supported by the National Natural Science Foundation of China; Project (20093219110035) supported by the Specialized Research Fund for the Doctoral Program of Higher Education of China
Corresponding author: DU Yu-lei; Tel: +86-25-84315159; E-mail: yldu_njust@mail.njust.edu.cn
DOI: 10.1016/S1003-6326(11)61228-4
Abstract: The first-principle calculations were performed to investigate the structural, mechanical, electronic and thermal properties of the binary ductile intermetallic compound CeAg with B2 (CsCl) structure. The calculated value of lattice constant a0 for CeAg with generalized gradient approximation is 3.713 ?, which is in better agreement with experimental data than local spin density approximation. The negative energy of formation implies that CeAg with B2 structure is thermodynamically stable phase. The greater separation between the d bands of Ce and Ag results in weaker bond hybridization of Ce d��Ag d, which prevents formation of directional covalent bonding. The three independent elastic constants (C11, C12 and C44) are derived and the bulk modulus, shear modulus, elastic modulus, anisotropy factor, and Poisson ratio are determined to be 57.6 GPa, 15.8 GPa, 43.4 GPa, 3.15 and 0.374, respectively. The elastic constants meet all the mechanical stability criteria. The value of Pugh��s criterion is 3.65. The ductility of CeAg is predicted if Pugh��s criterion is greater than 1.75. Furthermore, the variations of volume, bulk modulus, heat capacity, and thermal expansion coefficient with temperature and/or pressure were calculated and discussed.
