
���Ӷ���ѧģ��Pb30Ag70, Pb40Ag60��Pb60Ag40�Ͻ�����ѧ���ʵ��о�
����������ѧ���ұ����ҹ���ʵ����
��̨�����������ι�˾
ժ Ҫ��
�������ʵ�Ƕ��ԭ��ģ��ģ��Pb30Ag70, Pb40Ag60��Pb60Ag403�ֺϽ���ϵ�����¹���, ���ڼ���Ͻ���ϵ���ۼ��������, ����ģ��2981498 Kʱ��ϵ��������, �����õ���ͬ�¶��ºϽ���ϵ���������ܺ�ʣ������, Pb30Ag70, Pb40Ag60��Pb60Ag403�ֺϽ�Ĺ�ʣ�����ֱܷ�Ϊ1.26, 1.66��1.90 kJ, ��ʵ��ֵ���ϽϺ�, 3�ֺϽ�Ĺ�ʣ�����ܾ�Ϊ��ֵ, �Ͻ���ԭ�Ӽ�ƽ������ý�С��ͬʱ����Ͻ�Ľ���ܼ��γ��ܵ���������, ����õ�Pb-Ag�Ͻ�Ľ�������¶����߲��Ͻ���, ���Ͻ���ԭ�Ӽ�ƽ������ò��Ͻ��͡�Pb-Ag�Ͻ���γ���Ϊ��ֵ, �����Ͻ�Ϊ��ƫ����ϵ, �����¶ȵ�����, �Ͻ��γ��ܵľ���ֵ���Ͻ���������0, �����жϺϽ�������������ƫ��̶Ȳ��Ͻ���, �Ͻ���ϵ��������������, �Ͻ���Pb����Խ��, �Ͻ������������ƫ��̶�ԽС, ͨ�����γ��ܵļ�����Զ������жϺϽ������������ƫ��̶ȡ�
�ؼ��ʣ�
���Ӷ���ѧ;��������;���ʵ�Ƕ��ԭ�ӷ���;
��ͼ����ţ� TG111.3
����飺���, ͨѶ��ϵ��, (E-mail:kgyb2005@126.com) ;
�ո����ڣ�2009-12-25
����������Ȼ��ѧ�����������ϻ��� (U0837604) ������Ŀ;
Molecular Dynamics Simulation on Thermodynamic Properties of Pb30Ag70, Pb40Ag60 and Pb60Ag40 Alloy
Abstract��
The heating processes of Pb30Ag70, Pb40Ag60 and Pb60Ag40 alloys were simulated with generalized embedded atom method to calculate microscopic and macroscopic properties.The enthalpy of formation at 2981498 K was simulated at first, then free energy of formation and excess free energy were obtained.The excess free energies of Pb30Ag70, Pb40Ag60 and Pb60Ag40 were 1.26, 1.66 and 1.90 kJ, respectively, this agreed well with the experimental values.The excess free energies of three alloys were all positive, so the average atomic interaction was slight.The energy functions, such as cohesive energy and formation energy were calculated, and cohesive energy of Pb-Ag alloy decreased with the increase of the temperature, indicating the decrease of average atomic interaction.Formation energy of Pb-Ag alloy was positive, so the alloy showed positive deviation.With the temperature increasing, absolute value of formation energy approached to zero, and the alloy approached to ideal melt.The more the lead content, the smaller the deviation degree.The calculation of formation energy could describe the deviation degree between the actual alloy and the ideal melt quantitatively.
Keyword��
molecular dynamics (MD) ;energy functions;generalized embedded atom method;
Received�� 2009-12-25
���ЧӦʹ�Ͻ����������������ѧ���ʷ���һ���̶ȵ�ƫ��, �Ӷ�ʹ�Ͻ�ķ����ᴿ���̱�ø���, ��˶ԺϽ���ϵ����ѧ���ʵ��о�, �����Ϊ��Ҫ�� ���ڸ���ʵ��ĸ�����, Ŀǰ����������ϵ������ѧ���ʵõ����о�, ����������й����߶ԺϽ�����ѧ���ʵ����� ��ʮ���������о��߶���Һ����ѧ���ʵĽ���ʽ����˴����������뾭��ģ��, ��ȡ���˺ܴ�Ľ�չ
���������־���Ͱ뾭������Ʒ�չ�ܿ�, �ھ�ȷ�о����ϵ�����ѧ�ͽṹ���淢�Ӻܴ����á� �����ڽ������ϵĶ����ƿ��Է�Ϊ3��, ��Ƕ��ԭ�ӷ� (Embedded Atom Method-EAM)
1 ���Ӷ���ѧģ��
���Ӷ���ѧģ��Ĺؼ�����ѡ��ȷ������ģ�����������Ӽ�����á� ��Zhou��������ʵ�Ƕ��ԭ��ģ�� (GEAM��)
ʽ (1) ����ij (rij) ���������Ϊrij������ԭ��i, j֮��Ķ�������, Fi (��i) ��ʾԭ��i��Ƕ��ԭ����, ��i���Ա�ʾΪ
Ƕ��ԭ����Fi (��i) ���Ա�ʾ��:
Fni, Fi, Fo, ��Ϊģ�Ͳ���, ���ɽ���ܺ���ģ������õ�, ��eΪƽ��ʱ�ĵ����ܶȡ�
����������Ϊ:
re�������ԭ�Ӽ��ƽ�����, A, B, ��, ��Ϊ4��ģ�Ͳ���, ��, ��, m, n���йؽضϰ뾶�ĸ��Ӳ���, һ�������m=n=20�� ��=2��, ��/��=1.875
������Ͻ�ʱ, �Ͻ��Ʋ���
����?ab (r) Ϊa-b�Ͻ��������, fa (r) , fb (r) �ֱ���a��b��ֵĵ����ܶȺ���, ?aa (r) ��?bb (r) �ֱ���a��b��ֵ������ơ�
GEAM�ƺ����еIJ�������1��2��
���ķ��Ӷ���ѧģ��������500��ԭ�ӵ����������н���, �����漰��Pb, Ag����Ԫ�ؾ�Ϊ���������ṹ, ���ģ���������������Pb, Agԭ�������������ṹ�������, ģ������в���NPT (���µ�ѹ) ϵ��, ʹ�������Ա߽������� �Ͻ���ϵ���Ⱥ�����298 K, ��ԥ1��105��ʱ�䲽��, Ȼ����8.5��1012K��s-1������������1498 K, �����1498 Kʱ�ٳ�ԥ1��105��ʱ�䲽��, ʱ�䲽��ѡ��2��10-15s, ��ģ�������ѹǿ����0 Pa��
2 ���������
2.1�Ͻ������ʵļ���
ͼ1�Ǽ���õ�3�ֺϽ���ϵ��ͬ�¶��µ������ʡ� ��ͼ1�п��Կ�������õ�Pb30Ag70, Pb40Ag60��Pb60Ag40�Ͻ���۵��¶ȷֱ�Ϊ930, 925��830 K, ����
��1GEAM�Ƶ����ܲ��� (�����Ʋ���)
Table 1Potential parameters of GEAM (pair-potential)
Atom 1 |
Atom 2 | re, i | fe, i | ��i | ��i | Ai/eV | Bi/eV | ��i | Bj/eV |
Ag |
Ag | 2.892 | 1.106 | 7.945 | 4.237 | 0.266 | 0.386 | 0.425 | 0.386 |
Pb |
Pb | 3.450 | 0.648 | 8.468 | 4.516 | 0.135 | 0.203 | 0.426 | 0.203 |
Ag |
Pb | 2.892 | 1.106 | 7.945 | 4.237 | 0.266 | 0.386 | 0.425 | 0.203 |
| ��j | ��j | ��i | re, j | fe, j | ��j | ��j | Aj/eV | ||
Ag |
Ag | 0.425 | 0.851 | 0.851 | 2.892 | 1.106 | 7.945 | 4.237 | 0.266 |
Pb |
Pb | 0.426 | 0.852 | 0.852 | 3.450 | 0.648 | 8.468 | 4.516 | 0.135 |
Ag |
Pb | 0.426 | 0.852 | 0.851 | 3.450 | 0.648 | 8.468 | 4.516 | 0.135 |
��2GEAM�Ƶ����ܲ��� (�����Ʋ���)
Table 2Potential parameters of GEAM (many body potential)
Atom |
��e | Fno/eV | Fn1/eV | Fn2/eV | Fn3/eV | F0/eV | F1/eV | F2/eV | F3/eV | �� | Fe/eV |
Ag |
15.539 | 1.730 | -0.221 | 0.542 | -0.967 | 1.750 | 0.000 | 0.984 | 0.521 | 1.149 | 1.751 |
Pb |
8.907 | 1.420 | -0.229 | 0.630 | -0.561 | 1.440 | 0.000 | 0.921 | 0.109 | 1.172 | 1.440 |
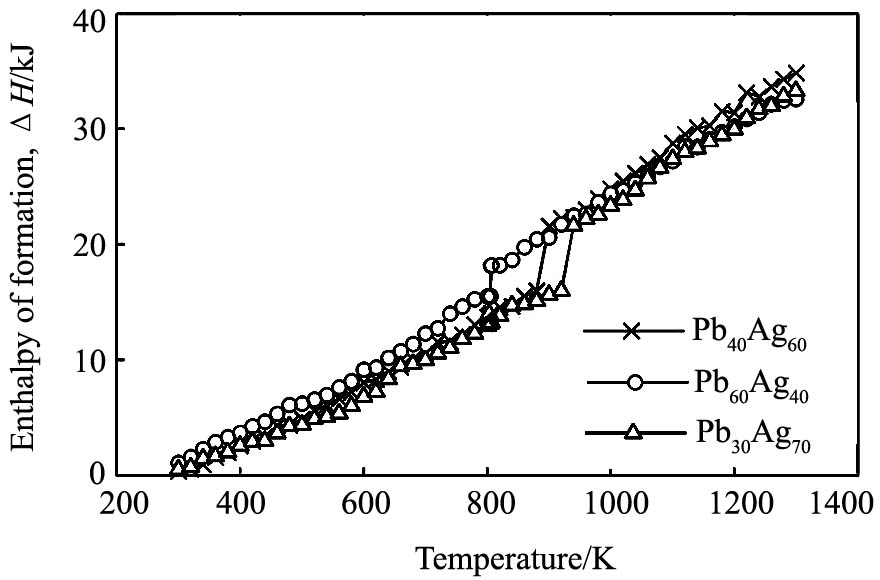
ͼ1 �Ͻ����������¶ȵı仯����
Fig.1 Temperature dependence of the enthalpy of formation
2.2�Ͻ��ʣ�����ܵļ���
���ݺϽ���ϵ������, ���Է���ؼ�����ϵ�����¹���������ѧ���ʡ� �Ͻ���ϵ�������������¶ȵĹ�ϵ�����ɹ�ʽ (6) ����ʾ��
��G (T) =��H (T) -T��S (T) (6)
�������۵�λ��ʱ, ��ϵ��Һ̬���������ܵ��ڹ�̬����������
��SLC (TM) =��H (TM) /TM (7)
���Ը���ʽ (7) , ���������¶��ºϽ���ϵ�������ء�
�� (8) ʽ���� (6) ʽ���ɼ������ͬ�¶��ºϽ���ϵ�����������ܡ�
��1273 Kʱ, ģ��õ���Pb30Ag70, Pb40Ag60��Pb60Ag40 3�ֺϽ���ϵ�����������ֱܷ�Ϊ: -94.68, -97.36��-104.16 kJ, ����
��ԪPb-Ag�Ͻ���ϵ�Ĺ�ʣ�����ܿ�����ʽ (8) ����õ���
��GE=��Gmix-��G
���Ц�GE�ǺϽ�Ĺ�ʣ������, ��Gmix�ǺϽ�Ļ��������, ��G
��Gmix=��G-xAg��GAg-xPb��GPb (10)
���Ц�GAg, ��GPbΪ��Ag, Pb������ͬ�¶��µ�����������, xAg, xPb�ֱ�ΪAg, Pb��ֵ�Ħ������, ��GΪ�Ͻ����������ܡ�
ͼ2Ϊ3�ֺϽ���ϵ��ͬ�¶��µĹ�ʣ������, ��ͼ2�п��Կ���1273 Kʱ����õ���Pb30Ag70, Pb40Ag60��Pb60Ag40 3�ֺϽ�Ĺ�ʣ�����ֱܷ�Ϊ: 1.26, 1.66��1.90 kJ, ʵ��ֵ
��ͼ2�п��Կ���3�ֺϽ�Ĺ�ʣ�����ܾ�Ϊ��ֵ, ˵���Ͻ���ԭ�Ӽ�ƽ������ý�С, ��ϵΪ��ƫ����ϵ; �����¶ȵ�����, �Ͻ�Ĺ�ʣ����������, �����¹����кϽ���ԭ�Ӽ�ƽ������ò��Ͻ���, ���¹��������ںϽ�ķ��롣
2.3�Ͻ����ܵļ���
�Ͻ���ϵ��ԭ�Ӽ�����û������ɺϽ�Ľ�������жϡ� ͼ3Ϊ����õ��IJ�ͬ�¶��½������ʼ��Ͻ����ܡ� ��298 KʱPb, Ag���ֽ������ʵļ�������Ϊ1.98, 2.81 eV, ʵ��ֵ
��ͼ3�п��Կ���:
(1) ��ͬ�¶���, ��Ǧ�Ľ����С�ڴ����Ľ����, ˵��Ǧԭ�Ӽ������ñ���ԭ�Ӽ�������С, Ǧԭ�ӽ���ԭ�Ӹ������γɹ���ԭ��, ����ϱ���ΪǦ������ѹ�����ߡ�
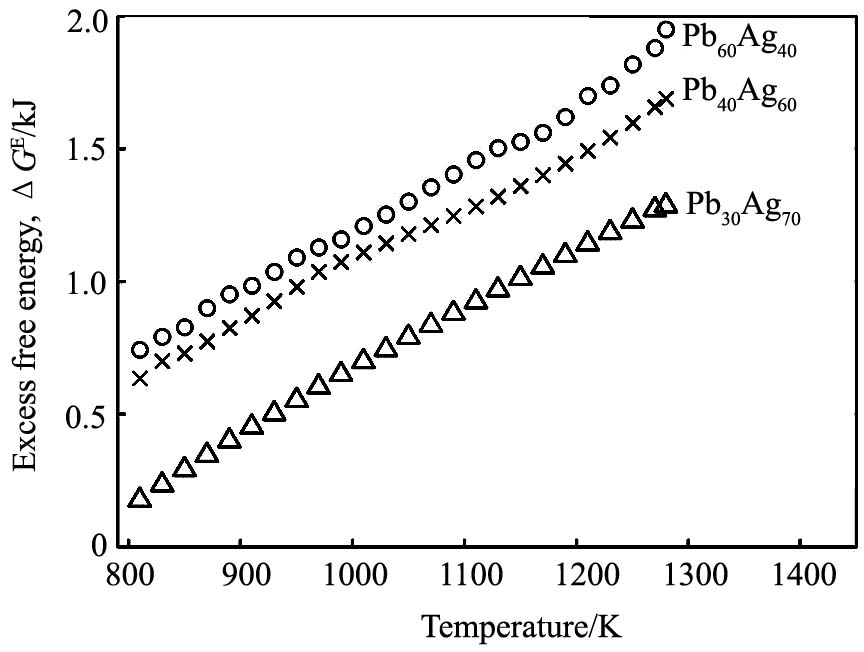
ͼ2 �Ͻ��ʣ���������¶��µı仯����
Fig.2 Temperature dependence of excess free energy

ͼ3 ��ͬ�¶��ºϽ�Ľ����
Fig.3 Temperature dependence of the cohesive energy
(2) Pb-Ag�Ͻ�Ľ�������¶����߲��Ͻ���, ���Ͻ���ԭ�Ӽ�ƽ������ò��Ͻ���, ˵���¶�Խ��, �Ͻ�Һ̬����Խ���γɹ����Ľ���ԭ��, ʹ����������ѹ���, ���������ڽ�����������
2.4�Ͻ��γ��ܵļ���
��ʣ�����ܿ����жϺϽ���ϵΪ��ƫ���ƫ��, �����ܱ����Ͻ���ϵ�����������ƫ��̶�, ��ͼ2�п��Կ���, �Ͻ���ϵ�Ĺ�ʣ���������¶ȵ����߲�������, ������˵���Ͻ������������ƫ��̶Ȳ�������, �෴�¶�Խ�ߺϽ���ϵ��������Һ��ƫ��̶�ԽС�� �Ͻ���γ��ܿ��Դ��۽Ƕ��������Ͻ����������������ƫ�����, �Ͻ��γɿ��Ա�ʾΪ:
Ef=-[EC (Pb-Ag) - (nPbEC (Pb) +nAgEC (Ag) ) / (nPb+nAg) ] (11)
ʽ (11) ��nPb, nAg�ֱ��ʾ�Ͻ���Ǧ�� �����ԭ�Ӹ���, EC (Pb-Ag) , Ec (Pb) , EC (Ag) �ֱ��ʾPb-Ag�Ͻ���ϵ�� ��Ǧ�� ������ϵ�Ľ���ܡ� ���Ͻ���ϵ���γ���Ϊ0ʱ, �Ͻ�Ϊ��������; ���Ͻ���ϵ���γ���Ϊ��ʱ, ˵���Ͻ���ԭ�Ӽ�ƽ������ý�С, ��ϵΪ��ƫ��; ���γ���Ϊ��ֵʱ, �Ͻ���ԭ�Ӽ�ƽ������ýϴ�, ��ϵΪ��ƫ� �Ͻ��γ��ܵľ���ֵԽ��, ����ʵ�ʺϽ������������ƫ��̶�Խ�� ͼ4Ϊ�Ͻ���ϵ���γ������¶ȵı仯���ߡ�
��ͼ4�п��Կ���Pb-Ag�Ͻ���ϵ���γ���Ϊ��ֵ�� �������¶���3�ֺϽ��Ϊ��ƫ����ϵ, �����¶ȵ�����, �Ͻ��γ��ܵľ���ֵ���Ͻ���������0, �����жϺϽ�������������ƫ��̶Ȳ��Ͻ���, �Ͻ���ϵ������������������, 3�ֺϽ���Pb����Խ��, �Ͻ������������ƫ��̶�ԽС, ���Ͻ��������

ͼ4 �Ͻ��γ������¶ȵı仯����
Fig.4 Temperature dependence of formation energy
3 �� ��
1. ������3�ֺϽ����������, ���Կ�������ֵ��ʵ��ֵ�Ǻ�, ˵��GEAM��������Pb-Ag�Ͻ���ϵ����ѧ���ʵ�ģ�⡣
2. 3�ֺϽ�Ĺ�ʣ�����ܾ�Ϊ��ֵ, ˵���Ͻ���ԭ�Ӽ�ƽ������ý�С, ��ϵΪ��ƫ����ϵ�� �����¶ȵ�����, �Ͻ�Ĺ�ʣ����������, ˵�����¹����кϽ���ԭ��֮��ƽ������ò��Ͻ���, ���¹��������ںϽ�ķ��롣
3. ��ϵ�Ľ�������¶ȵ����߲��Ͻ���, �����Ͻ���ԭ�Ӽ�ƽ������ò��Ͻ���, ������¹��������ںϽ�ķ��롣
4. ����õ��ĺϽ��γ��ܿ��Զ���������ʵ�ʺϽ������������ƫ��̶ȡ�
�����
[15] Johnson R A.Analytic nearest-neighbor modle for fcc metals[J].Phys.Rev.B, 1988, 37:3924.
[18] Smith C J.Metals Reference Book[M].London:Butterworrd, 1976.
