
���±�ţ�1004-0609(2010)05-0914-09
Sn�Ͻ�MgZn2�༰Mg2Sn��
�ṹ�ȶ��Եĵ�һԭ���о�
�ܵ���1�����ٻ�2���Ÿ�ȫ2���� ƽ2�� ����ˮ2
(1. ���ϴ�ѧ ���������Ƚ������������ص�ʵ���ң���ɳ 410082��
2. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ, ��ɳ 410082)
ժ Ҫ�����û����ܶȷ�������CASTEP��DMol�������������ӺϽ��γ��ȡ�����ܡ�����ѧ���ܺ͵��ӽṹ�ȷ��棬�о�Sn�Ͻ�MgZn2�༰Mg2Sn��Ľṹ�ȶ��ԣ�̽��Sn�Ͻ���ZA62þ�Ͻ�������ܵĻ����������������Sn��Al�ֱ��û�ZA62þ�Ͻ���MgZn2���Zn(��)��Zn(II)ԭ��ʱ����Sn��Al�û���MgZn2����Zn(��)ԭ�����γ��ȶ���MgZn2������ṹ����Sn��MgZn2���еĹ��������ޣ���Ͻ��γɵĹ�����ṹ��ȣ����ȶ��Ա�δ�Ͻ�ʱ�������������ĵڶ�������仯����Mg2Sn�Ľṹ��MgZn2�ĸ��ȶ�������ͬ�¶�������ѧ���ܵļ������������Ͻ���ϵ���γ��˽ṹ�ȶ���ǿ��Mg2Sn����ṹ�ȶ������¶�373��473 K�ķ�Χ�ڲ������¶ȵ����߶���ʧ���Ա�MgZn2�ĸߣ�����ZA62þ�Ͻ���ϵ���γ��˸����ȶ��Ե�Mg2Sn�࣬Sn�Ͻ�������ZA62þ�Ͻ�������ܵ���ߡ�����̬�ܶȺ�Mulliken����ռ�����ķ��������������MgZn2��Mg2AlZn3��Mg2SnZn3��������ȣ����ȶ���ǿ��Mg2Sn���γɵ���Ҫԭ������Mg2Sn ��ϵ�д���ǿ�ҵ����Ӽ��빲�ۼ��Ĺ�ͬ���á�
�ؼ��ʣ�
ZA62�Ͻ���MgZn2����Mg2Sn�����ṹ�ȶ���������ѧ��������һ��ԭ����Sn�Ͻ���
��ͼ����ţ�TG 146.2���� ���ױ�־�룺A
First-principle study on structural stability of
Sn alloying MgZn2 phase and Mg2Sn phase
ZHOU Dian-wu1, XU Shao-hua2, ZHANG Fu-quan2, PENG Ping2, LIU Jin-shui2
(1. State Key Laboratory of Advanced Design and Manufacturing for Vehicle Body,
Hunan University, Changsha 410082, China;
2. School of Materials Science and Engineering, Hunan University, Changsha 410082, China)
Abstract: By CASTEP and DMol program based on the density functional theory, the heat of formation, the cohesive energy, the thermodynamic properties and the electronic structure of the alloying system were investigated to study the structural stability of Sn alloying MgZn2 phase and Mg2Sn phase and explain the mechanism of the influence of Sn alloying on improving the creep resistance properties of ZA62 magnesium alloy. The results show that the structure of these phases can exist and be stable when the Zn atoms at the I positions of the MgZn2 phase are substituted with Sn and Al, whereas, it is also found that Sn is little solved in MgZn2 phase. By comparing with the stable MgZn2 phase, it is found that the stability of MgZn2 phase is reduced with Sn addition, and the structure of intermetallic compound Mg2Sn is more stable than that of MgZn2 phase. By calculating the thermodynamic properties of different phases, it is found that the improved creep resistance properties of ZA62 magnesium alloy are caused by forming intermetallic compound Mg2Sn with higher structural stability which is not changed with the elevated temperature in the range of 373-473 K. The calculations of the density of states (DOS) and Mulliken electronic populations of the alloying system show that the form of Mg2Sn with the highest structural stability in ZA62 magnesium alloy with Sn addition attributes to the ionic bond and covalent bond in the bonding electron numbers compared with those of MgZn2 phase, Mg2AlZn3 and Mg2SnZn3 solid solutions.
Key words: ZA62 alloy; MgZn2 phase; Mg2Sn phase; structural stability; thermodynamic property; first-principle; Sn alloying
�������ѳ�Ϊ������ҵ��չ�ı�Ȼ���ƣ���ΪĿǰ����Ĺ��̽�������֮һ������Ϊ��21������ɫ���̲��ϡ���þ�Ͻ���������ҵ�ϵ�Ӧ�õõ��˷�չ�� Ȼ����þ�Ͻ������ѧ���ܶ����ǿ�ȵ���������㷺Ӧ�ã����������¶ȸ���150 ��Ŀ��������þ�Ͻ��Ϊ����þ�Ͻ��о�������ȵ㡣��������ҵӦ����㷺��AZ91��AM60þ�Ͻ��У����ڴ�������ѧ���ȶ���Mg17Al12�࣬�����������¶���150������ʱ��þ�Ͻ��ǿ�ȺͿ�������ܼ����½�����˲���Ӧ���ڸ��»������Ͻ������������Լ�����������Mg17Al12�������þ�Ͻ�������ܵ���Ҫ;��������ZA62þ�Ͻ���ϵ�г��������-Mg�⣬������ǿ����MgZn2�����۵�(863 K)����AZ91��AM60þ�Ͻ���Mg17Al12����۵�(722 K)�����AZ62þ�Ͻ���������ҵ���й�����Ӧ��ǰ����
����о���������þ�Ͻ��м����IV��V ��Ľ���Ԫ��(��Sn��Bi��Pb��Sb)����ߺϽ���ϵ�����º��¿��������[1]�����йغϽ����þ�Ͻ���¿�������ܵ�Ӱ�����Ŀǰ������������[2]�о���Sn�Ͻ�ZA62þ�Ͻ���֯�����ܵ�Ӱ�죬������ϵ�г��˦�-Mg��Mg-Zn�����仯�����⣬������Mg2Sn�࣬��������Al��Sn������Mg-Zn�����仯�����С����ںϽ����ZA62þ�Ͻ���¿�������ܵ�Ӱ�����ʮ�ָ��ӣ�Ŀǰ�����ױ���Ҳ����[3-4]������չ�ⷽ��������о���ָ������������������Ҫ���壬Ϊ�ˣ��������߲��û����ܶȷ������۵�CASTEP��DMol�������������ӺϽ��γ��ȡ�����ܡ�����ѧ��������ӽṹ�ȷ��棬�о�Sn�Ͻ�MgZn2�༰Mg2Sn��Ľṹ�ȶ��ԣ����ӵ��Ӳ�εĽǶȣ�����Sn�Ͻ����ZA62þ�Ͻ�������ܵ�Ӱ�������
1 ����ģ���뷽��
C14��MgZn2��ľ���ṹ��ͼ1(a)��ʾ��������a=b=5.16 ?��c=8.56 ?���ռ�ȺΪP63/mmc����߶Գ���ΪD6h4��������ԭ������Ϊ12��ԭ������Ϊ��+4 Mg��(1/3��2/3��z), (1/3��2/3��1/2-z), z=1/16=0.062��+2 Zn(��)��(0��0��0)��(0��0��1/2)��+6 Zn(��)��(x��2x��1/4)��(-2x��-x��1/4)��(x��-x��1/4), x=-1/6=-0.170����Sn�Ͻ�ZA62þ�Ͻ���ϵʱ��Al��Sn������MgZn2���γɹ����壬�����¼��裺1) Al��Snԭ���滻MgZn2���е�Znԭ��Ϊͬ��ԭ���滻��2) �滻Znԭ�Ӻ�Al��Snԭ�ӵ�ԭ��λ�ô�����Ӧ�ľ���ѧλ�ã�3) �滻���Ŀռ�Ⱥ���䡣�����������ǣ��ֱ���2��Al��2��Sn�û�MgZn2�����е�2��Zn(��)����Sn�����������ӣ���6��Snԭ���û�6��Zn(��)ԭ�ӣ��û�����Ӧ�õ�Mg2AlZn3��Mg2SnZn3��MgSn2����ģ�ͣ��ֱ���ͼ1(b)��1(c)��1(d)��ʾ��������C1��Mg2Sn��ľ���ṹ��ͼ1(e)��ʾ��������a=b=c=6.69 ?���ռ�ȺΪFm3m����߶Գ���ΪOh5��������ԭ������Ϊ12��ԭ������Ϊ��+4 Sn��(0��0��0)��+8 Mg��(1/4��1/4��1/4)���ڼ�����������ģ�͵���������ӽṹʱ�����û����ܶȷ�������(Cambridge serial total energy package��CASTEP)�ij���������[5]�����������������ܡ������ܺͽ��������������֡����������ܲ��ù����ݶȽ���(GGA)�е�Perdew-Burke-Ernzerhof��ʽ[6], ���������ܵļ�������С���Ŀ��ٸ���Ҷ�任(FFT)�����Ͻ��У����ö������������г�ԥ�ij���(Ultrasoft)����[7]��Ϊƽ�沨������������Ǣ����(SCF)�������м��㡣������SCF����ʱ�����ý��BFGS(Broyden-Flecher- Goldfarb-
Shanno)�����ݶȷ�����Pulay�ܶȻ�Ϸ� ��[8-9]�������ӳ�ԥ���Ż�ʱ����ϵ������������ֵΪ5.0��10-6 eV/atom, ÿ��ԭ���ϵ�������0.01 eV/?, ����ƫ��С��5.0��10-4 ?��Ӧ��ƫ��С��0.02 GPa�����е����ܼ���ʱ�����ܽضϵ�Ϊ330.0 eV��FFT����Ϊ12��12��12������0.04 nm-1��K�ռ䣻����DMol������е�Dynamicsģ�����Ͻ���ϵ�ڲ�ͬ�¶��µ�����ѧ���ܡ�ѡȡ�������ϵ��(NVT)��ʱ�䲽��ȡ1.0 fs����ģ��ʱ��ȡ0.01 ps�����ӽ��������ܺ�������GGA���Ƶ�BLYP��ʽ[6]���ƺ���ȡȫ����λ�ƣ����Ӳ��������ô�d�����˫��ֵ��(DNP) ������FFT����Ϊ3��3��3������Smearing energy������������������
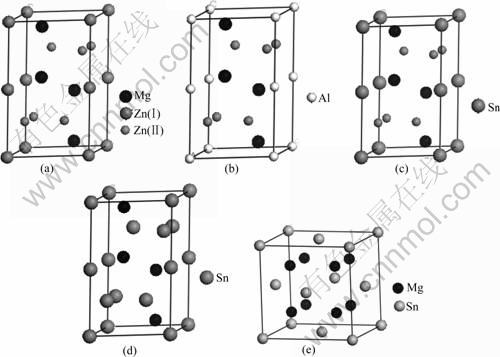
ͼ1 �����ṹ
Fig.1 Modes of MgZn2 (a), Mg2AlZn3 (b), Mg2SnZn3 (c), MgSn2 (d) and Mg2Sn (e)
2 �����������
����ʽ(1)������Sn�Ͻ�ǰ����MgZn2����ģ��ƽ��ÿ��ԭ�ӵĺϽ��γ���(?H)[10-11]��

���㾧̬��ԭ������ʱ����������㾧����������ͬ���ƺ�����Mg��Zn��Al��Sn���嵥ԭ�������ļ���ֵ�ֱ�Ϊ-977.87��-1 716.79��-57.24��-97.98 eV������õ���Sn�Ͻ�ǰ����MgZn2����ģ�͵ĺϽ��γ�����ͼ2��ʾ����ͼ2�ɼ���MgZn2��ĺϽ��γ���Ϊ-12.95 kJ/mol����ʵ��ֵ(-10.90 kJ/mol)[12]������ֵ(-8. 00 kJ/mol) [13]�ӽ���Al��Sn�û�MgZn2����Zn(��)ԭ�ӣ��Ͻ���ϵ�ĺϽ��γ���Ϊ���壬����Sn��Al�û�MgZn2����Zn(��)ԭ�����γ��ȶ���MgZn2������ṹ[15]����Snȫ��
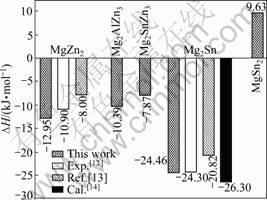
ͼ2 �Ͻ�MgZn2��Mg2AlZn3��Mg2SnZn3��Mg2Sn��MgSn2����γ���(��H)
Fig.2 Formation heats (��H) of MgZn2, Mg2AlZn3, Mg2SnZn3, Mg2Sn and MgSn2 phases
�û�MgZn2����Zn(��)��Zn(��) ԭ��ʱ���Ͻ���ϵ�ĺϽ��γ���ȴΪ�����������γɵ�MgZn2������ṹ���ȶ���Sn��MgZn2�еĹ��������ޣ�����������ʱ�����γɵڶ�������仯���
���[2]��ZA62þ�Ͻ����Sn�Ͻ�������ϵ�д���Mg2Sn�ࡣEDS�ķ�������(��ͼ3)����ɫ����״����Mg��ZnԪ��Ϊ������������Al��Sn��Al��Sn������Mg-Zn�����仯�����У����紦����

ͼ3 Sn�Ͻ�ZA62�Ͻ��SEM��
Fig.3 SEM image of as-cast ZA62 alloy after alloying with Sn
״����Mg��SnԪ��Ϊ������������Al��Zn�����ֿ�����ΪMg2Sn��ʵ��֤ʵ�������������ó��Ľ��[2]���������ۼ�����ʵ�������������߲���ʽ(2)��������仯����Mg2Sn�ĺϽ��γ���(?��H)��

Mg2Sn��ĺϽ��γ���Ϊ-24.46 kJ/mol����ʵ��ֵ(-24.30 kJ/mol)[16]�Ǻϣ�������ֵ(-20.82 kJ/mol[13]��-26.30 kJ/mol[14])Ҳ�ȽϽӽ�������������˵����Al��Sn������MgZn2�࣬���γ��ȶ��Ĺ�����ṹ��������Sn�Ͻ���������MgZn2��Ľṹ�ȶ����½�����Sn�Ͻ���ϵ�����Ľ����仯����Mg2Sn�ͺϽ�ǰ��MgZn2��ṹ���ȶ����ڡ�
�ֱ����ʽ(3)��ʽ(4)����Al��Sn�ֱ��û�MgZn2����Zn(��)��Zn(��)ԭ�ӣ������仯����Mg2Sn�Ľ����(E��coh)[11]��

Mg��Zn��Al��Sn����ԭ�������ļ���ֵ�ֱ�Ϊ-976.39��-1 715.56��-53.46��-93.87 eV������õ���MgZn2��Sn�Ͻ�ǰ����Ľ������ͼ4��ʾ����ͼ4�ɼ���MgZn2��Ľ����Ϊ89.056 kJ/mol����ʵ��ֵ(142.5 kJ/mol)������ֵ(139.6 kJ/mol)[12]�ӽ���Sn��Al�ֱ��û�MgZn2����Zn(��)ԭ���γ��ȶ���MgZn2������ṹ
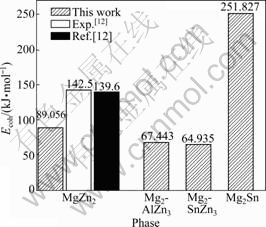
ͼ4 MgZn2��Mg2AlZn3��Mg2SnZn3��Mg2Sn�Ľ����(Ecoh)
Fig.4 Cohesive energies (Ecoh) of MgZn2, Mg2AlZn3, Mg2SnZn3 and Mg2Sn phases
Mg2SnZn3��Mg2AlZn3�Լ������仯����MgZn2��Mg2Sn��������Ӧ��ϵ�Ľ��������Ϊ��64.935��67.443��89.056��251.827 kJ/mol�����ھ���Ľ��ǿ��ͨ���ý���ܱ�ʾ[17]��������ܾ��ǽ�����ԭ�ӽ��Ϊ�������ͷŵ�������Ҳ���ǰѾ���ֽ�ɵ���ԭ������Ҫ���Ĺ��������Խ���γɵľ���Խ�ȶ�[18]����ˣ�Sn�û�Zn(��)ԭ��ʱ����ṹ��ȶ��������Al�û�MgZn2����Zn(��)ԭ��ʱ�ľ��壬�ٴ���MgZn2�࣬�����ȶ�����Mg2Sn�ࡣ
2.2�ڵļ�������������̬(��0 K) ʱ��Sn�û�Zn(��)ԭ��ʱ����ṹ��ȶ��������Al�û�MgZn2����Zn(��)ԭ�ӣ��ٴ���MgZn2�࣬�����ȶ�����Mg2Sn������ZA62þ�Ͻ��м���Ԫ��Sn��ʵ���¶Ȳ���Ϊ0 K��Ϊ�����¶ȶԽṹ�ȶ��Ե�Ӱ�죬�������߽�һ��������ϵ�ڲ�ͬ�¶���(298��573 K)������ѧ���ܡ�
������ϵ����ѧ������ѭ��������ѧͳ�ƹ�ʽ����ϵ��(H)������¶��µ�Gibbs������(G)�ֱ����ʽ(5)��(6)���м��㣺

Sn�Ͻ�MgZn2��������Mg2Sn��ϵ����(S)����(H) ��Gibbs������(G)�ļ������ֱ���ͼ5(a)��ͼ5(b)��ͼ6��ʾ����ͼ5�ɼ�������298 K (����)���ߵ�573 K(300 ��)ʱ���Ͻ���ϵ���غ��ʶ�������ͼ6(a)�ɿ����������¶ȵ����ߣ������仯����Mg2Sn��MgZn2�Լ�������Mg2AlZn3��Mg2SnZn3��Gibbs�����ܾ����٣�����ϵ�ṹ�ȶ������¶ȵ����߾��������͡���һ������������Mg2AlZn3�������Gibbs��������С�������Mg2Sn�ģ��ٴ���MgZn2�ģ�Gibbs������������Mg2SnZn3�����塣�������Sn�Ͻ�MgZn2��������ϵ�Ľṹ�ȶ��ԣ������¶ȵ����ߣ�������С�仯����Mg2AlZn3��������ϵ�Ľṹ�ȶ������¶����ߺ��ɲ���Mg2Sn��MgZn2�ȶ���Ϊ�����Ƕ��ȶ�����Mg2Sn��MgZn2��������Mg2SnZn3�Ľṹ���ȶ��Ե�ǿ��˳��ȴ�������¶ȵ����߶������仯��
���[2]�о����֣���ZA62þ�Ͻ��м���Ԫ��Sn�������˽����仯����Mg2Sn��2.1�ڵļ�����������Mg2Sn���ֽ����仯����Ľṹ�ȶ���2.2 ��

ͼ5 Sn�Ͻ�MgZn2��������ϵ��Mg2Sn�ڲ�ͬ�¶��µ�������
Fig.5 Entropies (S) (a) and enthalpies (H) (b) of Mg2Sn and MgZn2 phases at different temperatures with and without Sn addition
�ļ�������������ṹ����Ӧ�Ͻ�Mg2AlZn3��Mg2SnZn3�������Լ�MgZn2�Ľṹ���ȶ����ڴ˽�һ��������373��473 K(��100��200 ��)��Χ��Mg2Sn��MgZn2�Ľṹ�ȶ��������ͼ6(b)��ʾΪMgZn2��Mg2Sn��373��473 Kʱ��Gibbs�����ܣ�Mg2Sn��Gibbs�����ܱ�MgZn2�ĵ͡�����ZA62þ�Ͻ���ϵ�γ��˸����ȶ��Ե�Mg2Sn�࣬��ṹ�ȶ��Բ������¶ȵ����߶���ʧ���ұ�MgZn2�ĸߣ�Sn�Ͻ�������ZA62þ�Ͻ�Ŀ�������ܵ���ߡ�

ͼ6 Sn�Ͻ�MgZn2��������ϵ��Mg2Sn�ڲ�ͬ�¶��µ�Gibbs������
Fig.6 Gibbs free energies of Mg2Sn and MgZn2 phases at different temperatures with and without Sn addition
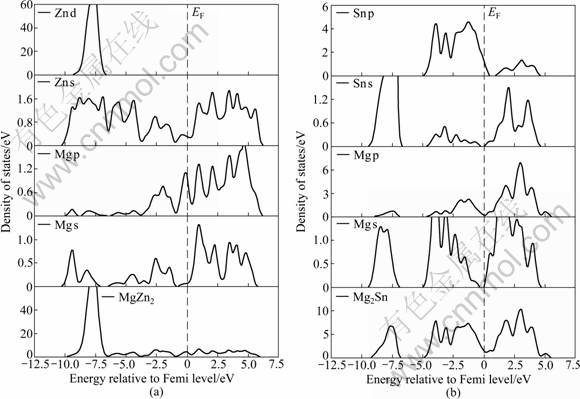
��2.3�ڵ�����ѧ���ܵļ�������֪��Sn�Ͻ���ǿZA62þ�Ͻ�Ŀ�������ܵ���Ҫԭ��������ϵ���γ��˲����¶����߶���ʧ��ǿ�Ľṹ���ȶ��Ե�Mg2Sn��MgZn2��������Mg2SnZn3��Mg2AlZn3��Ϊ��һ��������ṹ�ȶ��ĵ��ӻ��ƣ�������������MgZn2��Mg2AlZn3��Mg2SnZn3��Mg2Sn����̬�ܶ���ֲ�̬�ܶ�(��ͼ7)���������֣�MgZn2��0��-10 eV�ķ�Χ�ڣ��ɼ�������Ҫ��Mg s��Mg p��Zn s��
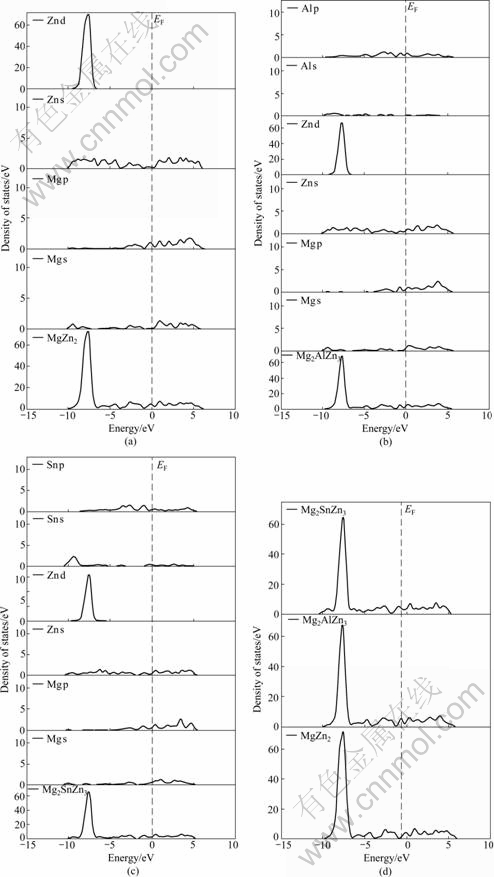
ͼ7 MgZn2��Mg2AlZn3�Լ�Mg2SnZn3��̬�ܶȺ���̬�ܶ�
Fig.7 Densities of states for MgZn2 (a), Mg2AlZn3 (b) and Mg2SnZn3 (c) and total density (d)
Zn d�ļ۵��ӹ���(��ͼ7(a))��Al������MgZn2���γ�Mg2AlZn3������(��ͼ7(b))����0��-10 eV�ķ�Χ�ڣ�Mg2AlZn3���������Ҫ�ɼ����ӳ�Mg s��Mg p��Zn s��Zn d�ļ۵��ӹ����⣬����һ��������Al s��Al p�۵��ӵĹ��ף���Sn������MgZn2���γ�Mg2SnZn3������(��ͼ7(c))����0��-10 eV��Χ�ڣ�Mg2SnZn3���������Ҫ�ɼ����ӳ�Mg s��Mg p��Zn s��Zn d�ļ۵��ӹ����⣬����һ��������Sn s��Sn p�ļ۵��ӹ��ס���һ���Ƚ�ͼ7(d)������MgZn2����Ҫ�ɼ���ֲ���0��-10 eV�ķ�Χ�ڣ��ɼ���ĸ߶�Ϊ71.596 6 electron/eV��Al������MgZn2�У��ֲ���0��-10 eV�ķ�Χ��Mg2AlZn3��Ҫ�ɼ���ĸ߶ȼ�С��Ϊ66.654 4 electron/eV����Sn������MgZn2�У��ֲ���0��-10 eV�ķ�Χ�ڣ�Mg2SnZn3��Ҫ�ɼ���ĸ߶Ƚ�һ����С��Ϊ62.536 8 electron/eV����MgZn2����̬�ܶ���ȣ�Al��Sn������MgZn2�У��ֱ��γɵ�Mg2AlZn3��Mg2SnZn3����0��-10 eV�ķ�Χ�ڣ��ɼ����������١�һ���棬�ɼ����������ٱ�����۵��Ӽ�����ü�������һ���棬���ٵijɼ�����λ�ڵ��ܼ���ʹ����ϵ��ṹ��ø��Ӳ��ȶ�����ˣ�MgZn2��ṹ�ȶ�����Al��Sn������MgZn2���У�������Ľṹ����MgZn2����ȶ�[18-19]��
Ϊ�˱Ƚ�Mg2Sn��MgZn2�Ľṹ�ȶ��ԣ���һ��������Mg2Sn��MgZn2����ģ�͵�̬�ܶ�(��ͼ8)�����ֶ���MgZn2��Mg2Sn���Գɼ��й����ӵ���������Ҫ������0��-10 eV�ķ�Χ�ڣ��ֱ���Դ��Mg s��Mg p��Zn s��Zn d�ļ۵��ӹ���(��ͼ8(a))�Լ�Mg s��Mg p��Sn s��Sn p�ļ۵��ӹ���(��ͼ8(b))����һ������ͼ8(a)���Կ���������MgZn2�Ͻ𣬵��ӹ���Ĺ����ӻ���Ҫ�ǽ�����Zn s-Mg s�ӻ�������Mg2Sn��Sn��sp̬��Mg��sp̬�����ӻ����ڷ����ܼ������γ��˽Ͽ��Ĵ�϶(��ͼ8(b))�����MgZn2��Mg2Sn�в����ӻ��ĵ���̬�������࣬�ӻ�ǿ��Ҳ��Ӧ��ǿ��
��1����ΪMgZn2��Mg2Sn��Mulliken����ռ�����ļ��������������֣�����MgZn2����Mgԭ����Znԭ�Ӳ����˵��ת�ƣ���ϵ��ת�Ƶ��������ԼΪ4.08(1.02��4)������Mg2Sn����Mgԭ����Snԭ�Ӳ����˵��ת�ƣ�ת�Ƶ������Ϊ4.72(0.59��8)�������������2�ֽ����仯�����ϵ�д��ڵ����Ӽ����ô�ǿ������˳��Ϊ��Mg2Sn��MgZn2��
��2�ֽ����仯�����̬�ܶ���Mulliken����ռ�����ļ�������֪��Mg2Sn�Ľṹ�ȶ�������MgZn2�ģ�������ϵ��ǿ�ҵ����Ӽ��빲�ۼ���ͬ���õĽ����
ͼ8 MgZn2�� Mg2Sn��̬�ܶ�
Fig.8 Densities of states of MgZn2 (a) and Mg2Sn (b)
��1 MgZn2��Mg2Sn��Mulliken����ռ����
Table 1 Mulliken electronic populations of MgZn2 and Mg2Sn

4 ����
1) �����ܶȷ�������CASTEP��DMol��������������Sn�Ͻ�MgZn2�༰Mg2Sn��ĺϽ��γ��ȡ�����ܡ�����ѧ��������ӽṹ��
2) ��Sn��Al�ֱ��û�ZA62þ�Ͻ�MgZn2����Zn(��)��Zn(��)ԭ��ʱ����Sn��Al�û���MgZn2����Zn(��)ԭ�����γ��ȶ���MgZn2������ṹ��Sn��MgZn2���й��������ޡ�
3) Mg2AlZn3��Mg2SnZn3������ṹ���ȶ��Ա�MgZn2�������������ڶ�������仯����Mg2Sn�Ľṹ��MgZn2�Ľṹ���ȶ���
4) Sn�Ͻ�������ZA62þ�Ͻ�ĸ��¿����������ߵ���Ҫԭ�Ͻ���ϵ���γ��˽ṹ�ȶ���ǿ��Mg2Sn�࣬��ṹ�ȶ��Բ������¶ȵ����߶���������373��473 K(100��200 ��)�ķ�Χ�ڣ���ṹ�ȶ����Ա�MgZn2��ĸߡ�
5) ��MgZn2��Mg2AlZn3��Mg2SnZn3��������ȣ����ȶ���ǿ��Mg2Sn����γɣ���Ҫԭ������Mg2Sn��ϵ��ǿ�ҵ����Ӽ��빲�ۼ���ͬ���õĽ����
REFERENCES
[1] ������, ������, Ԭ����. Sn ��þ�Ͻ�����֯����ѧ���ܵ�Ӱ��[J]. �й���ɫ����ѧ��, 1999, 9(1): 55-60.
SUN Yang-shan, WEN Kun-zhong, YUAN Guang-yin. Effects of Sn addition on microstructure and mechanical properties of magnesium alloys [J]. The Chinese Journal of Nonferrous Metals, 1999, 9(1): 55-60.
[2] �� �, �Ÿ�ȫ, �¼���, ����ǿ, ����ϲ. ���� ZA62 �Ͻ�����֯����ѧ���ܵ�Ӱ��[J]. ���켼��, 2006, 27(4): 378-381.
LIAO Kun, ZHANG Fu-quan, CHEN Ji-hua, ZOU Min-qiang, LIU Tian-xi. Effects of tin on microstructure and mechanical properties of ZA62 alloys [J]. Foundry Technology, 2006, 27(4): 378-381.
[3] �Ź�Ӣ, �� ��, ������, ���Ų�. Bi, Sb�Ͻ�AZ91þ�Ͻ���֯������Ӱ������о�[J]. ����ѧ��, 2005, 54(11): 5288-5292.
ZHANG Guo-ying, ZHANG Hui, FANG Ge-liang, LI Yi-cai. A study on the mechanism of the influence of Bi, Sb alloying on microstructure and properties of AZ91 magnesium alloy [J]. Acta Physica Sinica, 2005, 54(11): 5288-5292.
[4] DU Wen-wen, SUN Yang-shan, MIN Xue-gang, XU F, WU D Y. Influence of Ca addition on valence electron structure of Mg17Al12 [J]. Chinese Nonferrous Metals, 2003, 13(6): 1247-1280.
[5] SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14: 2717-2743.
[6] MARLO M, MILMAN V. Density-functional study of bulk and surface properties of titanium nitride using different exchange-correlation functionals [J]. Phys Rev B, 2000, 62: 2899-2907.
[7] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism [J]. Phys Rev B, 1990, 41: 7892-7895.
[8] HAMMER B, HANSEN L B, NORKOV J K. Improved adsorption energetics within density-functional theory using revised Perdew-Burke-Ernzerh of functionals [J]. Phys Rev B, 1999, 59: 7413-7421.
[9] FRANSCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calculations [J]. J Phys: Condens Matter, 1990, 2: 4395-4404.
[10] MEDVEDEVA M I, GORNOSTYREV Y N, NOVIKOV D L, MRYASOV V, FREEMAN A J. Ternary site preference energies, size misfits and solid solution hardening in NiAl and FeAl [J]. Acta Materialia, 1998, 46(10): 3433-3442.
[11] SAHU B R. Electronic structure and bonding of ultralight LiMg [J]. Mater Sci Eng B, 1997, 49(1): 74-78.
[12] LI C, HOE J L, WU P. Empirical correlation between melting temperature and cohesive energy of binary laves phases [J]. J Phys Chem Solids, 2003, 64(2): 201-212.
[13] ZHANG H, SHANG S L, SAAL J E, SAENGDEEJING A, WANG Y, CHEN L Q, LIU Z K. Enthalpies of formation of magnesium compounds from first-principles calculations [J]. Intermetallics, 2009, 17(11): 878-885.
[14] ANSARA I, DINSDALE A T, RAND M H. Thermodynamic database for light metal alloys [M]. Brussels: European Commission, 1998: 368.
[15] SONG Y, GUO Z X , YANG R, LI D. First principles study of site substitution of ternary elements in NiAl [J]. Acta Materialia, 2001, 49(9): 1647-1654.
[16] ZUBOV V I, TRETIAKOV N P. TEIXEIRA RABELO J N, SANCHEZ ORTIZ J F. Calculations of the thermal expansion, cohesive energy and thermodynamic stability of a van der Waals crystal-fullerene C60 [J]. Phys Lett A, 1995, 198(5/6): 470-471.
[17] ISHII Y, FUJIWARA T. Electronic structures and cohesion mechanism of Cd-based quasicrystals [J]. Non-cryst Solids, 2002, 312/314(12): 494-497.
[18] FU C L,WANG X D, YE Y Y, HO K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principles calculation[J]. Intermetallics, 1999, 7(2): 179-184.
[19] NYL?N J, GARC?A F J, MOSEL B D, P?TTGEN R, H?USSERMANN U. Structure relationships, phase stability and bonding of compounds PdSnn (n=2, 3, 4) [J]. Solid State Sci, 2004, 6(1): 147-155.
(�༭ �� ��)
������Ŀ����������ʿ��ר�����(�½�ʦ)����������Ŀ(200805321032)�����ϴ�ѧ���������Ƚ������������ص�ʵ���������о�����������Ŀ(60870005)
�ո����ڣ�2009-09-27�������ڣ�2009-12-30
ͨ�����ߣ��ܵ��䣬�����ڣ���ʿ���绰��13017297124��E-mail: ZDWe_mail@yahoo.com.cn
