
���±�ţ�1004-0609(2014)02-0343-08
Mg-Er�����仯�����ȶ�������ӽṹ�ĵ�һ��ԭ���о�
���ľ�1��������1����ϣ��1����־��2��������3
(1. �Ͼ����պ����ѧ ���Ͽ�ѧ�뼼��ѧԺ���Ͼ� 210016��
2. ɽ�����ֱ���������˾������ 273300��
3. �Ϻ���ͨ��ѧ ���Ͽ�ѧ�빤��ѧԺ���Ϻ� 200240)
ժ Ҫ��
ժ Ҫ�����û����ܶȷ������۵ĵ�һ��ԭ�����Ʒ����Ż�Mg-Er�Ͻ���ϵ��MgEr��Mg2Er��Mg24Er5��3�ֽ����仯����Ľṹģ�ͣ�ͨ���γ��ȡ�����ܺ͵��ӽṹ�ļ�������˻�������ȶ������侧��ṹ��������ϵ�����������3��Mg-Er�����仯������γ��Ⱥͽ���ܾ�Ϊ��ֵ����������γ��������ȶ��Ծ����Ż�������Er�����Ľ��Ͷ����͡��ڷ����ܼ����ܼ�����Mg��3s��2p�����Er��4f��5d��������ص��������˹���ӻ����ڷ����ܼ����ܼ�����Mg��2p�����Er��5d���Ҳ�����������ӻ������Ż�������Er�����Ľ��ͣ���������ƽ��ÿ��ԭ���ڷ����ܼ����ܼ����ijɼ����������٣���������ȶ��Խ��͡���Mg��Erԭ����Χ���д����ĵ�ɴ��ڣ��ʵ��͵Ľ�����������Mg��Er֮��ĵ�����ֻ�в����ص������紦��ɵĻ��䲻��Mg-Er�����仯����ļۼ���Ͼ��н��������ۼ������ԣ����н�����ռ������λ��Mg��Erԭ�ӵĵ��ת�����滯������Er�������Ͷ����٣�������Ĺ��ۼ��Խ��ͣ��ȶ����½���
�ؼ��ʣ�
Mg-Er�����仯��������һ��ԭ�����ȶ��������ӽṹ��
��ͼ����ţ�TB331 �� �� ���ױ�־�룺A
First-principles study on structural stabilities and electronic structures of Mg-Er intermetallic compounds
WANG Wen-jing1, LIU Zi-li1, LIU Xi-qin1, ZHANG Zhi-dong2, WANG Qu-dong3
(1. School of Material Science and Technology, Nanjing University of Aeronautics and Astronautics,
Nanjing 210016, China;
2. Shandong Menwo Transmission Co., Ltd., Linyi 273300, China;
3. School of Materials Science and Engineering, Shanghai Jiao Tong University, Shanghai 200240, China)
Abstract: Using the first-principle pseudopotential plane wave (PPW) method based on the density functional theory, structural optimization was conducted on MgEr, Mg2Er and Mg24Er5 intermetallic compounds of the binary Mg-Er alloys, and the internal relations of the stability and crystal structure of the Mg-Er intermetallic compounds were analyzed by calculating the formation heat, binding energy and electronic structure. The results show that in Mg-Er alloys, the formation heat and binding energy of three intermetallic compounds are all negative, and the alloying ability and structural stability of Mg-Er compounds are in decline with decreasing the content of Er. In low-energy region of Fermi level, the energy band is mainly dominated by hybridization of 4f and 5d orbits of Er with the 2p and 3s orbits of Mg, while in high-energy region of the Fermi level, those bonds are mainly contributed by electrons of 5d orbits of Er and 2p orbits of Mg. As the Er content decreases, the average quantities of bonding electron of each atom in low-energy region of Fermi level drops, which results in the weakened interaction among valence electrons and the reduced stability. There are a large number of charges around Mg and Er, suggesting the characteristics of typical metal bond. Meanwhile, Mg and Er share some charges to form covalent bond, while the distortion of the charge at the junction is little. Therefore, the valence bond of Mg-Er intermetallic compound has a characteristic of the duality wherein the metal bond predominates. With decreasing the Er content, the amount of charge transfer between Mg and Er atoms reduces gradually, which leads to the decrement of the proportion of covalent bond and the structural stability.
Key words: Mg-Er intermetallic compounds; first-principle; structural stability; electronic structure
��Ϊ����Ĺ��̽������ϣ�þ�Ͻ������������ѧ���ܺ����õ������ԡ������ӹ��ԡ������ԡ��������Լ���ǿ�ĵ���������������ڻ��յ�һϵ���ŵ㣬�ں��պ��졢������ͨѶ��ҵ�ϻ���˹㷺��Ӧ�á�ϡ��Ԫ��RE�����þ�Ͻ���ѧ���ܵ���Ҫ�Ͻ�Ԫ�أ�RE��Mg���нϸߵĹ����¶�(552~595 ��)��Nd��Ce��Ga��Er��ϡ��ԭ�Ӿ�����Mgԭ�ӽ���γɸ��Ӿ���ṹ�Ľ����仯�����Щ�����仯������ɢ�ֲ��ںϽ�����У����˺ܺõ�����ǿ����ϸ��ǿ�����ã��ܹ��������þ�Ͻ��������ѧ���ܡ��������ܺ����칤������[1-4]��
Mg-RE�����仯������RE��Mgԭ�ӵļۼ���Ͼ��н��������ۼ������ԣ�ֱ��Ӱ���˽����仯�������ѧ���ܺ��ȶ��ԣ������еĽ�����ռ������λʱ�������ԱȽϺã��ȶ��Խϲ��֮�������Խϲ�ȶ��ԽϺá����õ���������ص��о���������ԭ�ӳ߶��϶Ի�����ijɼ���ʽ�����о����������Ը��õ��˽�Mg-RE��Ԫ���������ѧ���ʡ��ȶ������侧��ṹ�����ӽṹ��������ϵ������Mg-RE�����仯����ĵ��������о������ѿ�չ�˺ܶ࣬��Ҫ����Nd��Ce��Ga��La��ϡ���ϡ�WANG��[5]���õ�һ��ԭ������ϵͳ�о���Mg-Nd�����仯����Ľṹ�ȶ��ԣ�֤��Mg-Nd�Ͻ��г���״̬�µ�����ǿ����Ӧ����Mg41Nd5������Mg12Nd�ࡣYANG��[6]������Ե�[7]�о���Mg-Ce��ϵ��MgCe��Mg2Ce��Mg3Ce��3�ֽ����仯����ĵ��ӽṹ�͵������ʣ��ó��˻�������ȶ��ԡ����������뾧��ṹ��������ϵ���ơ�Mg-Ga�Ͻ���ϵ�д����������仯�����࣬����Mg5Ga2��Mg2Ga��MgGa��O-MgGa2(����)��Mg2Ga5���ȶ��࣬��H-MgGa2(����)�������ࡣGAO��[8]ͨ�����ۼ���ó�6��Mg-Ga�����仯����ĵ������ʺ�����ѧ���ܣ�Ϊ���ȶ��Ե��о��ṩ���·�����Mg-La�Ͻ���ϵ�д���5�ֽ����仯���MgLa��Mg2La��Mg3La��Mg17La2��Mg12La������Mg-La��������La�������½��������ܼ����ܼ�����ɼ����������٣��������γ��ܵľ���ֵ��С���ȶ��Խ���[9]��
Er������ϡ����������Mg�е������ܶ�(32.7%)Զ��������������ϡ��Ԫ�صģ���ˣ�Er��þ�Ͻ������Ӱ��Ч���ȳ��õ���ϡ��Ԫ�ظ�Ϊ��������Erþ�Ͻ���о��ܵ��˼���Ĺ�ע[10-13]�����ں�Erþ�Ͻ���о���Ҫ��������ѧ���ܺ���ʴ���ܷ��棬Ŀǰ��δ������Mg-Er�����仯����ĵ������۷�����о��������������߲��û����ܶȷ������۵ĵ�һ��ԭ�����㷽���Ż���MgEr��Mg2Er��Mg24Er5��3��Mg-Er�����仯����ľ���ṹ��ͨ���γ��ȡ�����ܵļ��㲢��ϵ��ӽṹ�����˽����仯������ȶ������۽ṹ��ԭ�Ӽ��Ϸ�ʽ֮��Ĺ�ϵ��
1 ģ������㷽��
1.1 ����ṹ��ģ��
����Mg-Er��Ԫ��ͼ��������Mg-Er�Ͻ��д���MgEr��Mg2Er��Mg24Er5��3�ֽ����仯�����3�ֽ����仯����ṹ�������1����[14-17]��MgEr�и�ԭ�ӵ�����ֱ����£�Er(0��0��0)��Mg(0.5��0.5��0.5)��Mg2Er�и�ԭ�ӵ�����ֱ����£�Er(0.333 3��0.666 7��0.063 0)��Mg(0��0��0)��(0.833 0��0.666 0��0.250 0)��Mg24Er5�и�ԭ�ӵ�����ֱ����£�Er(0��0��0)��(0.312 6��0.312 6��0.312 6)��Mg(0.355 7��0.355 7��0.032 4)��(0.092 7��0.092 7��0.279 1)��3�ֻ�����Ľṹģ�ͷֱ���ͼ1��ʾ��
1.2 ���㷽��
���û����ܶȷ������۵ij���������CASTEP (Cambridge serial total energy package)������3��ģ�͵���ؼ���[18]��ѡ������ݶȽ���GGA(Generalized gradient approximation)�µ�PBE(Perdew-Burke- Ernzerhof)��������������������[19]���۵����������Ƴ��ó������������죬��Ǣ������ʱ����Pulay�ܶȻ�Ϸ������ռ���ƽ�沨���������ֹ����EcutΪ380.0 eV��MgEr��Mg2Er����Brillouin����K��ȡΪ5��5��2��Mg24Er5��ȡΪ2��2��2����Ǣ������Ϊ1��10-6 eV/atom���ṹ�Ż�����ʱ����ϵ������������2��10-5 eV/atom��ÿ��ԭ���ϵ�������0.5 eV/nm������ƫ��Ϊ2��10-4 nm��Ӧ��ƫ��Ϊ0.1 GPa�����ǵ��������漰��ԭ�Ӱ���ϡ��ԭ��Er������������4f������Ӳ㣬��ģ�ͽ�����������صļ���ʱҪ��Er��f���Ӳ����Hubbard U����[20]��U��Ϊ6��
��1 Mg-Er�����仯����ṹ����[14-17]
Table 1 Structure parameters of Mg-Er intermetallic compounds[14-17]
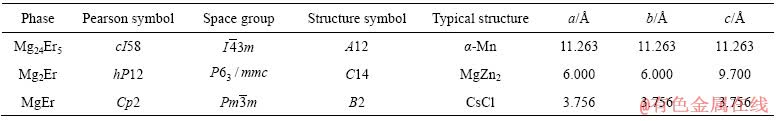
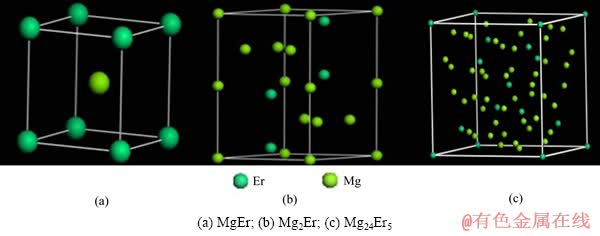
ͼ1 Mg-Er�����仯���ᄃ��ģ��
Fig. 1 Cell models of Mg-Er intermetallic compounds
�ڽ����仯������γ��Ⱥͽ���ܼ����н��漰��Mg��Er����ԭ�ӹ�̬�Լ�����̬ʱ����������2�г���Mg��Erԭ�ӵľ������͡���̬��ԭ����������ʱK��ͽض���Ecut�����á�����̬��ԭ����������ʱ�ض���Ecut�����á�����ԭ�ӹ�̬�Լ�����̬���������������̬��ԭ������ʱ������������Ϊ10  �ļ������ṹģ�ͣ�������ԭ������ģ�͵�����λ�ã�K����ΪGamma�㡣
�ļ������ṹģ�ͣ�������ԭ������ģ�͵�����λ�ã�K����ΪGamma�㡣
2 ������������
2.1 ������
��3����Ϊ��MgEr��Mg2Er��Mg24Er5���������ԭ��λ���Ż�������ƽ�⾧�����;�������Etot��MgEr��Mg2Er��Mg24Er5�ľ�����������[14-17]������ʵ��ֵС5%�������Ż��������ɿ���
2.2 ���������
�γ�����ָԭ���ɵ���״̬�γɻ�����ʱ�ͷŵ������������ڱ��������仯�����γɵ����׳̶ȣ����γ���Ϊ��ֵʱ�������ֵԽ��ʾ�˽����仯����Խ���γ�[21-23]��Mg-Er�����仯������γ���HΪ[24]
 (1)
(1)
ʽ�У�x��y�ֱ��ʾ�����仯���ᄃ���ṹ��Mg��Erԭ�ӵ�ԭ�Ӹ�����Etot��ʾ�����ᄃ���ṹ�Ż���ɺ����������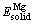 ��
��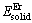 �ֱ��ʾ����Mg��Erԭ���ڹ�̬ʱ���������ڼ����̬��ԭ��������ʱ������������仯���ᄃ����������ͬ�ļ����������ֱ�Ϊ��Mg��Erԭ����Ӧ�ĵ����������м��㣬Ȼ����Ե������ܵ�ԭ�Ӹ��������õ���������Mg��Er�Ĺ�̬��ԭ��������Mgԭ����Erԭ�ӵĹ�̬��ԭ�������ֱ�Ϊ-973.951 eV/atom��-5 224.094 eV/atom(��2)����ϱ�3��3�ֻ�������������ݣ�����ʽ(1)����ó�MgEr��Mg2Er��Mg24Er5���γ��ȷֱ�Ϊ-1.386��-1.084��-0.214 eV/atom�����Կ�����3�ֻ�������γ��Ⱦ�Ϊ��ֵ������MgEr�γ��ȵľ���ֵ����γ�������ǿ��Mg2Er��֮��Mg24Er5���γ��������(��ͼ2(a))��
�ֱ��ʾ����Mg��Erԭ���ڹ�̬ʱ���������ڼ����̬��ԭ��������ʱ������������仯���ᄃ����������ͬ�ļ����������ֱ�Ϊ��Mg��Erԭ����Ӧ�ĵ����������м��㣬Ȼ����Ե������ܵ�ԭ�Ӹ��������õ���������Mg��Er�Ĺ�̬��ԭ��������Mgԭ����Erԭ�ӵĹ�̬��ԭ�������ֱ�Ϊ-973.951 eV/atom��-5 224.094 eV/atom(��2)����ϱ�3��3�ֻ�������������ݣ�����ʽ(1)����ó�MgEr��Mg2Er��Mg24Er5���γ��ȷֱ�Ϊ-1.386��-1.084��-0.214 eV/atom�����Կ�����3�ֻ�������γ��Ⱦ�Ϊ��ֵ������MgEr�γ��ȵľ���ֵ����γ�������ǿ��Mg2Er��֮��Mg24Er5���γ��������(��ͼ2(a))��
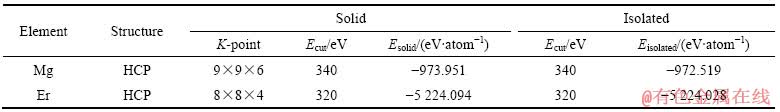
��2 ��̬������̬����ԭ�ӵ�����
Table 2 Energies of solid and isolated atom
��3 MgEr��Mg2Er��Mg24Er5��ƽ�⾧�����뾧������
Table 3 Equilibrium lattice constants and total energy of MgEr, Mg2Er and Mg24Er5

���������ǽ�����ԭ�ӽ��Ϊ�������ͷŵ�������Ҳ���ǰѾ���ֽ�ɵ���ԭ��ʱ��������Ĺ��������ǿ�ȡ��ṹ�ȶ�����������������أ��������Ϊ��ֵʱ�������ֵԽ�����γɵľ����Խ�ȶ�[21, 23, 25]��Mg-Er�����仯��������EcohΪ[24]
 (2)
(2)
ʽ�У� ��
��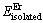 �ֱ��ʾMg��Er����ԭ�ӵ���������ϱ�2�еĹ���̬ԭ���������ݣ�MgEr��Mg2Er��Mg24Er5�Ľ���ܼ������ֱ�Ϊ-2.095��-2.060��-1.410 eV/atom(��ͼ2(b))��3�ֻ��������ܵľ���ֵ���Ż�������Er�����ļ��ٶ����ͣ�������Mg-Er�Ͻ�ϵͳ�У���Ȼ3�ֻ���������ȶ����ڣ�����������ȶ�������Er�����ļ��ٶ����͡����е��о������������ϡ��Ԫ��Gd��Dy��Tb��Mg�Ķ�Ԫ�Ͻ���ϵ�о�����3�ֽ����仯�����ࣺMgRE��Mg2RE��Mg24RE5�����ȶ��Ծ���ѭ���Ż�������ϡ��ԭ�Ӻ����Ľ��Ͷ����͵Ĺ��ɣ�ͬ��������ϡ��Ԫ�ص�Er��Mg�γɵĶ�Ԫ�����仯������ȶ��Թ�����������ϡ��Ԫ�ص���ͬ[26]��
�ֱ��ʾMg��Er����ԭ�ӵ���������ϱ�2�еĹ���̬ԭ���������ݣ�MgEr��Mg2Er��Mg24Er5�Ľ���ܼ������ֱ�Ϊ-2.095��-2.060��-1.410 eV/atom(��ͼ2(b))��3�ֻ��������ܵľ���ֵ���Ż�������Er�����ļ��ٶ����ͣ�������Mg-Er�Ͻ�ϵͳ�У���Ȼ3�ֻ���������ȶ����ڣ�����������ȶ�������Er�����ļ��ٶ����͡����е��о������������ϡ��Ԫ��Gd��Dy��Tb��Mg�Ķ�Ԫ�Ͻ���ϵ�о�����3�ֽ����仯�����ࣺMgRE��Mg2RE��Mg24RE5�����ȶ��Ծ���ѭ���Ż�������ϡ��ԭ�Ӻ����Ľ��Ͷ����͵Ĺ��ɣ�ͬ��������ϡ��Ԫ�ص�Er��Mg�γɵĶ�Ԫ�����仯������ȶ��Թ�����������ϡ��Ԫ�ص���ͬ[26]��
2.3 ̬�ܶ�

ͼ2 MgEr��Mg2Er��Mg24Er5���γ��Ⱥͽ����
Fig. 2 Heat of formation(a) and binding energy(b) of MgEr, Mg2Er and Mg24Er5
ͼ3��ʾΪ3��Mg-Er�����仯����ķֲ�̬�ܶ�(PDOS)����̬�ܶ�(TDOS)ͼ����3�ֻ��������̬�ܶ�ͼ���Կ����������ܼ����ĵ�������Ϊ�㣬˵����3�ֻ�����������ԵĽ������ԡ�3�ֻ��������̬�ܶ�ͼ�гɼ����ӵķֲ��������һ�£���Ҫ������4���������䣺-47~-49 eV��-43~-44 eV��-21~-23 eV��-6~0 eV��-47~-49 eV�۴����ĵ�����Ҫ��Er��6s������ף�-43~-44 eV�۴����ĵ�����Mg��2p������ף�-21~-23 eV�۴����ĵ�����Er��5p������ף���-6~0 eV�۴�����Mg��3s��2p�����Er��4f��5d��������ص���˵���ڸ���������˹���ӻ���Mg-Er�����仯�������Ҫ�ɼ���ֲ���-6~0 eV�ķ�Χ�ڡ�Mg��2p�����Er��4d����ڵ�����Ҳ��һ�������ӻ��������ܼ����ܼ�����ĵ�������Ҫ�ijɼ����ӣ��ڷ����ܼ���������ԭ���յ�Er 5d̬���ڵ��ת�Ʊ�ñ�����ռ��(��������Er 6s����������Er 4f)[27]�������Er 5d���ӶԽ����仯������ԭ�Ӽ�ijɼ�Ҳ�й��ס�

ͼ3 MgEr��Mg2Er��Mg24Er5�ķֲ�̬�ܶ�(PDOS)����̬�ܶ�(TDOS)ͼ
Fig. 3 Partial and total DOS of MgEr(a), Mg2Er(b) and Mg24Er5(c)
�״����ȵĴ�С�ܹ���ӳ�����Ӿ���̶ȵ�ǿ���͵��Ӳ���ɼ������Ĵ�С���״�����С�����Ӿ���̶�ǿ��ԭ�ӹ����չ���������Ӳμӳɼ�������С����֮������Ӿ���̶�����ԭ�ӹ����չ��ǿ�����Ӳμӳɼ�����������PDOSͼ�п��Կ�����Mg��s��p��������˷����渽���ĵ״����ȡ�Mg-Er 3�ֻ�����ĵ״��ܼ��ֱ���-6~12��-6~3��-6~1 eV֮�䡣�Ա�3�ֻ������TDOSͼ�����Կ��������Ż�������Er���������ӣ�3�ֻ������ڷ����ܼ������ĵ״���������������Ҫ�����ڷ����ܼ��ұ߸���������ȵ�������ˣ�ErԪ�صļ�������˵��Ӳμӳɼ���������������ķǽ����Ժ��ȶ�����Er���������߶���ǿ��
�ںϽ���ϵ�У��Ͻ�Ԫ�ص�ƽ���ɼ��������ܺܺõط�ӳ�Ͻ���ȶ��ԡ��ɼ�������Ҫλ�ڷ����ܼ����£����и���ĵ��Ӵ��ڽϵ�����ʱ���ɼ����������ӣ������ڵļ۵��ӵ��������ǿ��������ȶ��Խ�����[28]��ʹ�õ���̬�ܶȻ��ֵķ��������ƽ��ÿ��ԭ���ڷ����ܼ����ܼ����ijɼ����������Դ�Ϊ�оݣ������жϳ��Ͻ��и����������ȶ��Եĸ� ��[29-30]��Mg-Er�Ͻ���ϵ�У�MgEr��Mg2Er��Mg24Er5��ƽ��ÿ��ԭ���ڷ����ܼ����ܼ����ijɼ��������ֱ�Ϊ7.38��5.99��3.82�����Կ���������Er�����ļ��٣����������仯����ƽ��ÿ��ԭ���ڷ����ܼ����ܼ����ijɼ����������٣�3�ֻ�������ȶ�����Er�����Ľ�������С�ģ�������������ܼ���ó��Ľ���һ�¡�
2.4 ����ܶ����ֵ���ܶ�
ͼ4��ʾΪ3��Mg-Er�����仯����(110)��ĵ���ܶ�ͼ���ɵ���ܶ�ͼ���Կ�����ÿ��ԭ����Χ�ĵ�ɾ������ηֲ�����Mg��Erԭ����Χ���д����ĵ�ɴ��ڣ��ʵ��͵Ľ�����������
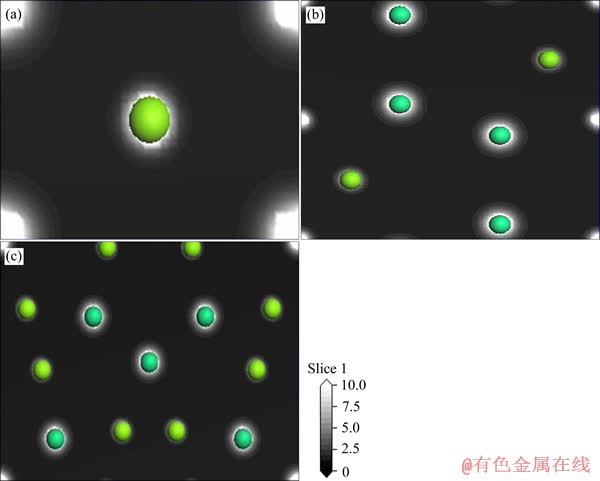
ͼ4 MgEr��Mg2Er��Mg24Er5��(110)�����ܶ�ͼ
Fig. 4 Charge densities on (110) plane of MgEr(a), Mg2Er(b) and Mg24Er5(c)

ͼ5 MgEr��Mg2Er��Mg24Er5��(110)���ֵ���ܶ�ͼ
Fig. 5 Charge densities difference on (110) plane of MgEr(a), Mg2Er(b) and Mg24Er5(c)
ͼ5��ʾΪ3�ֻ�����(110)��IJ�ֵ���ܶ�ͼ�������ܶȲ��ú졢�ס�����ʾ����ɫ��ʾ�õ�������ɫΪʧ��������ɫ���м�̬�����Կ�����3�ֻ������(110)���ϵ�Er��Mgԭ����Χ�����˵��ת�ƣ�Erԭ�Ӻ�Mgԭ��λ��Ϊʧ����������ܶȲ�Ϊ��ֵ��������֮��Ϊ�õ���������ܶȲ�Ϊ��ֵ������Mgԭ����Erԭ�Ӵ��ڹ��õ��ӣ�Mg-Erԭ�Ӽ��γ��˷����Խ�ǿ�Ĺ��ۼ�����ͼ3��̬�ܶȷ�����֪��Mg��Erԭ�Ӽ���������Ĺ���ӻ������ɲ�ֵ���ܶ�ͼ���Կ�����Mg��Er֮��ĵ�����ֻ�в����ص������紦��ɵĻ��䲻�����ԣ�Mg-Er�����仯����ļۼ���Ͼ��н��������ۼ������ԣ����н�����ռ������λ�����ۼ���ռ������С���Ա�MgEr��Mg2Er���ֻ�����IJ�ֵ���ܶ�ͼ���Կ���(��ͼ5)��MgEr��Mgԭ����Erԭ��֮��ĵ��ת��������Mg2Er����ԭ��֮��ĵ��ת�����������MgEr��Mg-Er���ۼ���ǿ�ȸ���Mg2Er�еġ����ۼ�ǿ��Խ�ߣ�������ȶ���Խ��[31]�����ԣ�MgEr���ȶ���Ҫ��Mg2Er�ĸߣ�������������ܺ�̬�ܶ����ý���һ�¡���Mg24Er5�IJ�ֵ���ܶ�ͼ��Erԭ���ܼ��ֲ��ĵط���ԭ�Ӽ���ת�����ϴ�˵������Щ���ۼ��ԱȽ�ǿ������Erԭ�ӷֲ����ٵ�����ԭ�Ӽ���ת�������٣�˵������Щ���ۼ��ԱȽ���������Mg24Er5������Erԭ�Ӻ����ϵͣ����ԣ�����ͬԭ������3��Mg-Er������ľ����У�Mg24Er5��Mg-Er���ۼ����������٣�����ĵ��ת������С���ȶ�����
3 ����
1) Mg-Er�����仯����MgEr��Mg2Er��Mg24Er5���γ��Ⱥͽ���ܾ�Ϊ��ֵ���������Ż�������Er�����Ľ��ͣ���������γ��������ȶ��Զ����͡�
2) ����Er�����Ľ��ͣ������仯����ƽ��ÿ��ԭ���ڷ����ܼ����ܼ�����ijɼ���������֮���٣������м۵��ӵ�����ü�������������ȶ��Խ��͡�
3) ��Mg��Erԭ����Χ�д����ĵ�ɴ��ڣ��ʵ��͵Ľ�����������Mg��Er֮����ڹ��õ��ӣ��γ��˷��ۼ��������紦��ɵĻ��䲻��Mg-Er�����仯����ļۼ���Ͼ��н��������ۼ������ԣ����н�����ռ������λ��Mg��Erԭ�ӵĵ��ת�����滯������Er�������Ͷ����٣�������Ĺ��ۼ��Խ��ͣ��ȶ����½���
REFERENCES
[1] WANG Qu-dong, LU Yi-zhen, ZENG Xiao-qin, DING Wen-jiang, ZHU Yan-ping. Effects of RE on microstructure and properties of AZ91 magnesium alloy[J]. Transactions of Nonferrous Metals Society of China, 2000, 10(2): 235-239.
[2] XIN Ren-long, LI Li, ZENG Ke, SONG Bo, LIU Qing. Structural examination of aging precipitation in a Mg-Y-Nd alloy at different temperatures[J]. Materials Characterization, 2011, 62(5): 535-539.
[3] ��ʫ��, κ����, �ֺ�ͬ, ����ʿ. �Ƽ�������ϡ����AZ91þ�Ͻ���̬��֯��Ӱ��[J]. �й���ɫ����ѧ��, 2001, 11(S2): 99-102.
ZHANG Shi-chang, WEI Bo-kang, LIN Han-tong, WANG Li-shi. Effect of yttrium and mischmetal on as-cast structure of AZ91 alloy[J]. The Chinese Journal of Nonferrous Metals, 2001, 11(S2): 99-102.
[4] ������, ��λά, �����, ��־��, �� ��, ������. Er����̬AZ91þ�Ͻ�����֯����ʴ���ܵ�Ӱ��[J]. �й���ɫ����ѧ��, 2009, 19(5): 847-853.
LIU Chu-ming, GE Wei-wei, LI Hui-zhong CHEN Zhi-yong, WANG Rong, GAO Yan-rui. Effect of Er on microstructure and corrosion resistance of AZ91 magnesium alloy[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(5): 847-853.
[5] WANG Can, HAN Pei-dei, ZHANG Lu, ZHANG Cai-li, XU Bing-she. First-principles study on the stabilities of the intermetallic compounds in Mg-Nd alloys[J]. Rare Metal Materials and Engineering, 2011, 40(4): 590-594.
[6] YANG Fang, WANG Ji-wei, KE Jiang-ling, PAN Zheng-gui, TANG Bi-yu. Elastic properties and electronic structures of Mg-Ce intermetallic compounds from first-principles calculations[J]. Physica Status Solidi B, 2011, 248(9): 2097-2102.
[7] �����, ������, ������, ������, �ܻ�Ӫ. Ce-Mg�Ͻ���ϵ������������ӽṹ�ĵ�һ��ԭ���о�[J]. ���ֵ��ӿƼ���ѧѧ��, 2012, 32(4): 339-344.
WANG Dian-wu, WANG Zhong-min, JI Zong-wei, HU Chao-hao, ZHOU Huai-ying. First-principle study of elastic properties and electronic structure of Ce-Mg alloys[J]. Journal of Guilin University of Electronic Technology, 2012, 32(4): 339-344.
[8] GAO Qian-nan, DU Yong, ZHAO Dong-dong, WANG Ai-jun, WANG Jiong, LIU Shu-hong, QU Yi-fang. Elastic, phonon and thermodynamic properties of Mg-Ga compounds from first-principles calculations[J]. Computer Coupling of Phase Diagrams and Thermochemistry, 2012, 37: 137-144.
[9] WANG Yu-fei, ZHANG Wei-bing, WANG Zhi-zhong, DENG Yong-he, YU Na, TANG Bi-yu, ZENG Xiao-qin, DING Wen-jing. First-principles study of structural stabilities and electronic characteristics of Mg-La intermetallic compounds[J]. Computational Materials Science, 2007, 41: 78-85.
[10] �����, ������, ���dž�, �����, ���Ͼ�. ��̬Mg-Zn-Er-Zr�Ͻ�����֯����ѧ���ܵ��о�[J]. �����ȴ�������, 2008, 37 (14) : 39-41.
QIU Cong-zhang, LIU Chu-ming, CHANG Ya-zhe, LI Hui-zhong, WANG Meng-jun. Research on microstructure and mechanical properties of Mg-Zn-Er-Zr alloy as-cast[J]. Material and Heat Treatment, 2008, 37(14): 39-41.
[11] �Ž���, ����ƽ, �� ��, �� ȫ. ��Er��ZM5þ�Ͻ�����֯����ʴ���ܵ�Ӱ[J]. ���켼��, 2012, 33(2): 160-163.
ZHANG Jin-tao, CHEN Ping-le, YIN Jian, ZHOU Quan. Influence of Er addition on microstructure and corrosion resistance of ZM5 magnesium alloy[J]. Foundry Technology, 2012, 33(2): 160-163.
[12] WANG Zhong-jun, XU Yang, ZHU Jing. Effects of erbium addition on the corrosion resistance and microstructure of AZ91 magnesium alloy[J]. Advanced Materials Research, 2011, 194/196: 1221-1224.
[13] �� ��, Ԭ����, ���곽, �����, ������. ϡ��Er��ZK21þ�Ͻ���֯��Ӱ��[J]. �����ȴ���ѧ��, 2011, 32(11): 94-98.
ZHANG Jing, YUAN Fu-qing, DOU Yu-chen, ZHANG Xu-feng, TANG Ai-tao. Effects of rare earth Er on microstructure of ZK21 alloy[J]. Transactions of Materials and Heat Treatment, 2011, 32(11): 94-98.
[14] BUSCHOW K H J. Magnetic properties of CsCl-type rare earth-magnesium compounds[J]. Journal of the Less Common Metal, 1973, 33(2): 239-244.
[15] KRYPYAKEVICH P I, EVDOKIMENKO V I, ZALUTSKII I I. Hexagonal Laves phases in the alloys of magnesium with rare-earth metals[J]. Reports of the Academy of Sciences of the Ukrainian SSR, 1964, 6: 766-769
[16]  W, BUSCHOW K H J. Neutron diffraction study on intermetallic Er5Mg24 and Tm5Mg24[J]. Materials Science Forum, 2004, 443: 263-266.
W, BUSCHOW K H J. Neutron diffraction study on intermetallic Er5Mg24 and Tm5Mg24[J]. Materials Science Forum, 2004, 443: 263-266.
[17] SACCONE A, DELFINO S, MACCIO D, FERRO R. Phase equilibria in the binary rare-earth alloys: The erbium-magnesium system[J]. Metallurgical Transactions A, 1992, 23(3): 1005-1012.
[18] SEGALL M D, LINDAN P L D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condens Matter, 2002, 14: 2717-2743.
[19] PERDEW J P, CHEVARY J A, VOSKO S H, JACKSON K A, PEDERSON M R, SINGH D J. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation[J]. Physical Chemistry B, 1992, 46: 6671-6687.
[20] ANISIMOV V I, ZAANEN J, ANDERSEN O K. Band theory and Mott insulators: Hubbard U instead of Stoner I[J]. Physical Review B, 1991, 44: 943-954.
[21] WU Meng-meng, WEN Li, TANG Bi-yu, PENG Li-ming, DING Wen-jiang. First-principles study of elastic and electronic properties of MgZn2 and ScZn2 phases in Mg-Sc-Zn alloy[J]. Journal of Alloys and Compounds, 2010, 506: 412-417.
[22] FU C L, WANG X D, YE Y Y, HO K M. Phase stability, bonding mechanism, and elastic constants of Mo5Si3 by first-principle[J]. Intermetallics, 1999, 7(2): 179-184.
[23] CHEN L��, PENG Ping, LI Gui-fa, LIU Jin-shui, HAN Shao-chang. First-principle calculation of point defective structures of B2-RuAl intermetallic compound[J]. Rare Metal Materials and Engineering, 2006, 35(7): 1065-1070.
[24] SAHU B R. Electronic structure and bonding of ultralight LiMg[J]. Materials Science and Engineering B, 1997, 49(1): 74-78.
[25] SONG Y, GUO Z X, YANG R, LI D. First principles study of site substitution of ternary elements in NiAl[J]. Acta Materialia, 2001, 49(9): 1647-1654.
[26] ������. ϡ��þ�Ͻ������ѧ���������Ե�����ģ��[D]. ��ɳ: ���ϴ�ѧ, 2007: 72-81.
WU Yu-rong. Theory simulation for thermodynamic and solid solution properties of eare magnesium alloys[D]. Changsha: Hunan University, 2007: 72-81.
[27] ������. ������d/f���ӵĻ�������ӽṹ�ĵ�һ��ԭ���о�[D]. �Ϻ�: ������ѧ, 2007: 75.
MA Lan-chun. First-principles studies on electronic structures of compounds containing local d/f electrons[D]. Shanghai: Fudan University, 2007: 75.
[28] GHOSH G. First-principles calculations of structural energetics of Cu-TM (TM=Ti, Zr, Hf) intermetallics[J]. Acta Materialia, 2007, 55: 3347-3374.
[29] YU R, HE L L, YE H Q. Effect of W on structural stability of TiAl intermetallics and the site preference of W[J]. Physical Review B, 2002, 65(18): 184102-184107.
[30]  ,
,  U. Structure relationships, phase stability and bonding of compounds PdSnn(n=2, 3, 4)[J]. Solid State Sciences, 2004, 6(1): 147-155.
U. Structure relationships, phase stability and bonding of compounds PdSnn(n=2, 3, 4)[J]. Solid State Sciences, 2004, 6(1): 147-155.
[31] �����, �� ��, �� ��, ������. Al-Sc�����仯����ĵ��ӽṹ���ȶ��Ժ�����ѧ����[J]. �й���ɫ����ѧ��, 2010, 20(5): 946-953.
LI Yan-feng, XU Hui, ZHANG Biao ZHANG Li-gang. Electronic structure, stability and thermodynamic properties of Al-Sc intermetallics compounds[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(5): 946-953.
(�༭ ������)
������Ŀ��ɽ��ʡ�������³ɹ�ת���ش�ר����Ŀ(2012ZHZX0601)
�ո����ڣ�2013-05-22�������ڣ�2013-11-22
ͨ�����ߣ������������ڣ���ʿ���绰��025-52112626��E-mail: liuzili@nuaa.edu.cn
