
J. Cent. South Univ. (2016) 23: 2173-2181
DOI: 10.1007/s11771-016-3274-y
First-principles study on electronic structure, magnetic and dielectric properties of Cr-doped Fe3C
YANG Jian-ping(�ƽ), CHEN Jin(�½�), LI Wei(��ΰ), HAN Pei-de(�����), GUO Li-na(������)
College of Materials Science and Engineering, Taiyuan University of Technology, Taiyuan 030024, China
 Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Abstract:
The first-principles calculations were performed to investigate the electronic structure, magnetic and dielectric properties of Cr-doped Fe3C, in comparison to those of pure Fe3C and Cr3C. The obtained results show that the thermodynamic stability of Cr- doped Fe3C becomes weaker in terms of the larger formation enthalpy, on the contrary, the metallicity and covalency are found to strengthen to some extent. The magnetic moments of Fe3C, Fe11CrC4(g), and Fe11CrC4(s) are respectively 21.36 ��B/cell, 16.92 ��B/cell, and 17.62 ��B/cell, and in Fe11CrC4(g) and Fe11CrC4(s), the Fe of Wyckoff positions of 8d and 4c is substituted by Cr. The local magnetic moment of Cr at 8d site is larger than that at 4c site in the doped structure, which is opposite to that of Fe. In low frequency band, the permittivity follows the ranking of Fe11CrC4(s)>Cr3C>Fe11CrC4(g)>Fe3C. Once exceeding a certain frequency, the sequence will be broken. Besides the electron transition, the polarization of atoms also makes a contribution to the dielectric properties.
Key words:
Cr-doped Fe3C; electronic structure; magnetic properties; dielectric properties��
1 Introduction
q-Fe3C (cementite) as a kind of fundamental carbide in Fe-C alloy system is the most classical and important phase existing in white cast iron and various of carbon steels, which plays a critical role for the strength and applicability of materials [1-3]. Several intrinsic properties of Fe3C-carbide have been studied by material metallurgy researchers. HALLSTEDT et al [4] reevaluated the Gibbs energy function of cementite from 0 K to 1000 K as well as the heat capacity using density functional theory (DFT). LV [5] calculated the mechanical, electronic and magnetic properties of ortho-Fe3C and hexa-Fe3C by first-principles method. In order to achieve a better mechanical performance for actual industrial demand, several alloying elements, primarily transition metals of Mn, Cr, Co, and Ni, generally are doped into the Fe3C carbide, and these addition agent can affect the structure, characteristics, morphology and growth kinetics of the Fe3C carbide. The previous groups have also investigated the electronic structure and formation enthalpy of (Fe, M)3C (M=Cr/Mn/Co/Ni), finding that Cr/Mn/Co/Ni doping all can improve the stability of cementite [6-7]. On account of the significant anisotropy of Fe3C as well as (Fe, M)3C, the surface structure and relevant physical properties of them also have been addressed in the theoretical side [7-10]. Compared to other cementite-type alloys, Cr-doped cementite, namely (Fe, Cr)3C, is the most common, extensively consisting of in sorts of stainless steel and other chrome steel. LV et al [6] and ZHOU [11] further studied the stability, magnetic property of (Fe, Cr)3C with different positions and contents in Cr doping through first principle calculation and obtained the favourable results. Regrettably, the influence of single Cr atom doping in different lattice sites on the above properties and electronic structure is still unclear.
(Fe, Cr)3C not only is being considered an important strengthening phase in steel alloy, but also still appears in metallurgical raw materials. In this work, it��s mainly indicated high-carbon ferrochrome (HCFC) studied by our groups. In the production of stainless steel, the decarburization of HCFC is regarded as the intermediate process, but it determines the yield and quality of stainless steel. Recently, the solid-phase decarburization of high-carbon ferrochrome powder (HCFCP) by microwave heating, instead of bulk materials derived from inevitably generating so-called skin-effect that intensively reflects microwave [12], has attracted major interest of researchers in the metallurgy fields in view of microwave showing simultaneous ordering of rapid warming, body heating, selective heating, especially no environmental pollution and easy for automatic control,importantly avoiding the generation of large amounts of toxic chromium slag [13]. As widely accepted by researchers, the dielectric properties of ferrochrome carbide in HCFCP can significantly take account for the temperature rising characteristic and reaction performance of HCFCP in the microwave field [14-15]. CHEN et al [14] discussed the intrinsic nature of (Cr, Fe)7C3, the main phase in HCFCP, based on density functional theory and indicated the importance of microstructure of (Cr, Fe)7C3 on microwave dielectric properties of HCFCP. Besides (Cr, Fe)7C3, CrFe also has been investigated to concern the magnetic property rather than dielectric properties as its remarkable metallicity, which contributes to microwave heating yet [16]. Unluckily as the unneglected phase in HCFC, the dielectric and magnetic properties of (Fe, Cr)3C are detailedly unknown all the time.
In terms of the shortage of previous research about (Fe, Cr)3C, the electronic structure, magnetic and dielectric properties of Cr-doped Fe3C will be synthetically discussed in our present work to give a better support and instruction for the theoretical research and practical production in the fields of metallurgy and material. Considering the limit of Cr additive in most steel alloys as well as the relatively low Cr content in (Fe, Cr)3C contained within HCFC, single Cr atom doping Fe3C in different lattice sites, namely Fe11CrC4, will be dramaticlly studied compared with pure Fe3C and Cr3C in this paper.
2 Crystal structure and computational details
Since the orthorhombic structure is extensively concerned by academia as well as meets our experimental finding by X-ray scattering techniques, the space group of Fe3C is determined as Pnma (S.G. No. 62). Every crystal cell includes four Fe3C units, where eight (four) iron atoms are located in the ����general���� (����special����) sites, namely Wyckoff positions of 8d (4c), and four carbon atoms are in the interstices [6-11]. When the Fe atoms of Fe3C in different sites are substituted by one Cr atom respectively, Fe11CrC4(g) and Fe11CrC4(s) (g and s, representing general and special) are obtained. It is noteworthy that the valence-electron configurations for Fe, Cr and C atoms are 3d64s2, 3p63d54s1 and 2s22p2, respectively. Although the differences between Fe and Cr on atomic radius couldn��t be neglected, the ideal Cr-doped Fe3C models for calculations are similar to the structure of Fe3C after geometry optimization, as shown in Fig. 1.
All of the density-functional-theory (DFT) calculations were accomplished using the Cambridge Sequential Total Energy Pack-age (CASTEP) with ageneralized gradient approximation (GGA+U) [17] combining the Perdew�CWang function (PW91) [18], to deal with the exchange and correlation energy of electrons. The electron�Cion interaction was described by the Vanderbilt ultrasoft pseudopotentials (USPPs) [19]. To ensure the validity and accuracy, the calculations were constructed with kinetic energy cutoff of 350.0 eV and a k-point grid of 5��4��6, using Monkhorst�CPack in the first irreducible Brillouin zone [6]. The crystal structures were fully relaxed with BFGS (Broydene- Fletchere-Goldarbe-Shanno) scheme, where both of the atomic positions and cell constants were optimized simultaneously [11]. As the ground states of Fe3C and Fe11CrC4 are ferromagnetic, all of the calculations were carried out by spin polarized method. The total energy was well-converged to less than 2��10-6 eV per atom and the forces were reduced to 0.05 eV/ -1 per atom.
-1 per atom.
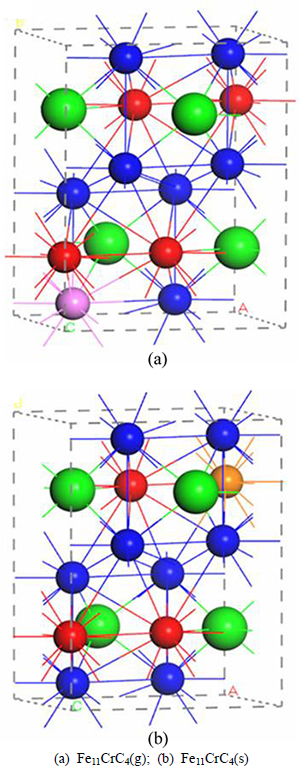
Fig. 1 Constructed models: (The small blue balls, small red balls and big green balls respectively represent Fe(g) atoms, Fe(s) atoms and C atoms; The pink and orange balls represent Cr atoms substituting for Fe atoms in the ��general�� and ��special�� sites, respectively)
3 Results and discussion
3.1 Structural optimization and thermodynamic stability
The ground state properties of Fe3C, Fe11CrC4(g), Fe11CrC4(s), and Cr3C are obtained from its total energy, which is calculated as a function of volume. By a series of iterations until it is fitted to the Murnaghan equation of state [20], the equilibrium lattice constants are list in Table 1. To verify the accuracy and credibility of results in our calculation, the experimental data and some related theoretical calculations with different research methods are also listed in Table 1. Besides equilibrium lattice constant, cohesive energy and formation enthalpy of them are displayed in table as well, which are calculated by the equations shown in previous literature [11].
From Table 1, it can be seen the results calculated in our present work such as lattice parameters, Etot, Ecoh, and ��rHm are in a good agreement with the experimental values and other theoretical predictions. Obviously our calculated method performs the rationality and reliability, and these results can give enough support to further predict the properties of Fe3C-type carbides. While in Table 1, the negative ��rHm of Fe3C obtained by the local density approximation (LDA) method suggests an incorrect prediction for bulk thermodynamic stability of compounds, since it��s known widely that Fe3C belongs to a sort of meta-stable structure. In addition, two types of Cr-doped carbide are both the meta-stable phases as well, and their greater ��rHm values mean the lower stability than Fe3C, which is stemed from more highlighted metallic anti-bonding that refers to the chemical bonds that have the negative overlap population [26] in Cr- doped Fe3C as given in Table 3. With respect to Cr3C, it reveals the best stability than other carbides with the following sequence of Cr3C>Fe3C>Fe11CrC4(g)>Fe11CrC4(s). It is however interesting that Cr3C can be hardly discovered in nature, which may be derived from most other carbides in Cr-C system having the lower formation enthalpy than Cr3C [25]. Comparing two types of Cr-doped Fe3C, the volume of Fe11CrC4(s) is slightly larger than Fe11CrC4(g) on account of Cr atom in special site possessing high exposure beyond the surface.
3.2 Chemical bond and magnetic properties
The chemical bonds of above carbides are discussed and compared in this part. Figure 2 shows the partial densities of states (PDOS) of Fe3C, Fe11CrC4(g), Fe11CrC4(s), and Cr3C. Apparently, the PDOSs of these carbides all can be divided into three regions: the lowest valence band, the upper valence band and the conduction unoccupied states. For Fe3C and Cr-doped Fe3C, the lowest valence band ranging from -14 to -11.5 eV is mostly composed of orbits for C 2s and Fe(Cr) 4s; The upper valence band can be approximatively separated into two parts: The first one is ranging from -8 to about -5 eV, which is constituted with the hybridization of C 2p and Fe(Cr) 3d orbits, reflecting the feature of covalent; The second one, mainly formed by Fe(Cr) 3d orbits with a few C 2p orbits, is from -5 eV up to Fermi level. Moreover, 3d orbits of metal atoms also have the great contributions to the conduction band, especially those of up-spin in Cr atoms as well as down-spin of Fe atoms. When Fe is replaced by Cr, the changes of PDOS mainly occur in 3d orbits of Fe(Cr), implying the importance of 3d orbits for bonding. In spite of the PDOS of Cr3C moving along the x-axis towards the higher energy, the better stability of Cr3C is due to the energy of most electrons placed in s and p orbits of Cr atoms below -50 eV (Not shown in figures). Similarly, there also exists Cr 3d hybridization with C 2p in PDOS of Cr3C. No energy gap near the Fermi level as well as the finite DOS at the Fermi level can be observed in all carbides, which indicates the metallic nature. Further, the metallicity (fm) of four kinds of carbides can be estimated by [25]
 (1)
(1)
where nm and ne are thermal excited electrons and valence electron density of the cell unit respectively; kB is the Boltzmann constant; T is the thermodynamic temperature; Df is the DOS value of cell unit at the Fermi level; Vcell is the cell volume; and N is the total number of the valance electrons in the cell. The calculated results showing in Table 2 list the order of the metallicity,Fe11CrC4(s)>Fe11CrC4(g)>Fe3C>Cr3C. Therefore, Cr doping improves the metallicity of Fe3C to some extent. Furthermore, the tiny difference of fm for Fe11CrC4(s) and Fe11CrC4(g) demonstrates there is a weak relation between the metallicity and doping site.
Table 1 Calculated and experimentally measured lattice constants, cell volume (Vcell), cell total energy (Etot), cohesive energy (Ecoh) and formation enthalpy (��rHm) of Fe3C, Fe11CrC4, and Cr3C
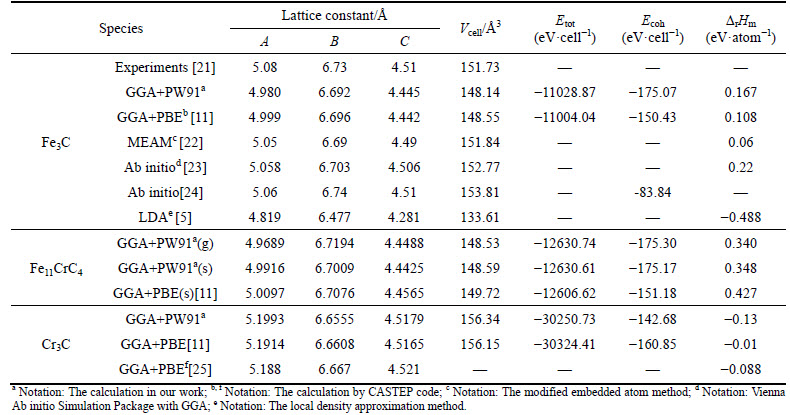
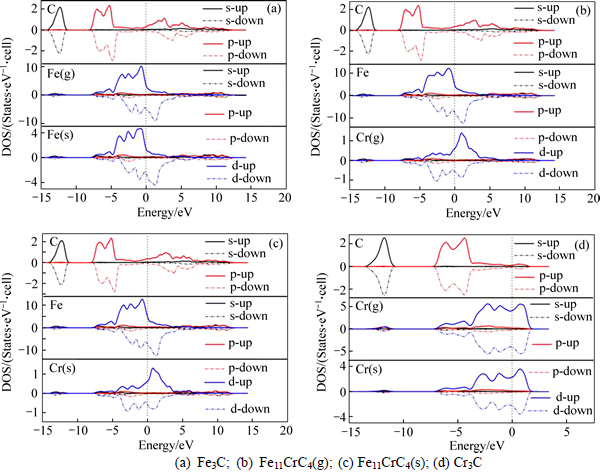
Fig. 2 Calculated site- and spin-polarized partial densities (PDOS) of states of (Fe, Cr)3C carbides:
Table 2 Calculated cell volume (Vcell), DOS at the Fermi level (Df), metallicity (fm) and total number of valance electrons (N) of Fe3C, Fe11CrC4, and Cr3C
Besides the covalent and metallicity, above carbides also possess the nature of ionicity, and it can be predicted that Cr3C has the stronger ionicity for the larger energy gap between the lowest valence and the upper valence band compared to other carbides [10]. On the basis of electronic differential density (generally defined as the electron density difference between the isolated atoms and the bonding states with the same atomic configurations) for  plane shown in Fig. 3, the darker red of Cr than Fe implies more charge transfer to C atoms, verifying a remarkable electropositivity of Cr.By the further study for Fe11CrC4(s) and Fe11CrC4(g), it is observed that the electron clouds around Fe atoms prefer to be perpendicular to the ��surface�� of Cr atoms, which may be originated from the noteworthy attractive interaction to electrons due to their strong electropositivity. In addition, the darker red of Cr(s) atoms in the special site against Cr(g) atoms in the general site reveals a more positive valence, namely a larger contribution of electrons for cell.
plane shown in Fig. 3, the darker red of Cr than Fe implies more charge transfer to C atoms, verifying a remarkable electropositivity of Cr.By the further study for Fe11CrC4(s) and Fe11CrC4(g), it is observed that the electron clouds around Fe atoms prefer to be perpendicular to the ��surface�� of Cr atoms, which may be originated from the noteworthy attractive interaction to electrons due to their strong electropositivity. In addition, the darker red of Cr(s) atoms in the special site against Cr(g) atoms in the general site reveals a more positive valence, namely a larger contribution of electrons for cell.
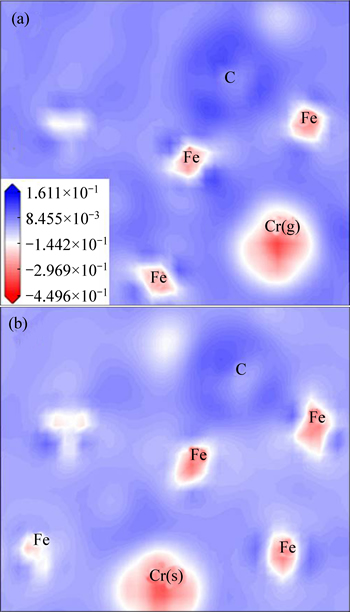
Fig. 3 Electron density difference distribution Fe11CrC4(g) (a) and Fe11CrC4(s) (b) at 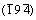 plane
plane
By virtue of Mulliken population analysis, it can gain an insight into the characteristic of covalent bonds. From Table 3, it is clear that C��Fe(s) represents the stronger covalent bonds in comparison to C��Fe(g) in Fe3C, due to the shorter bond length and the larger overlap population. For C��Cr bond in Cr-doped Fe3C and Cr3C, C��Cr(s) also possesses the stronger covalent bonds than C��Cr(g). Discussing the average bond length and overlap population of C��Fe for Cr-doped Fe3C and Fe3C, the sequence of the bond length of C��Fe is Fe11CrC4(s)> Fe3C>Fe11CrC4(g) with the order of the overlap population following Fe11CrC4(g)>Fe3C> Fe11CrC4(s). In regard to C��M bonds (the average value of all covalent bonds composed of metal atoms and C atoms in cell unit) of Fe11CrC4(g) and Fe11CrC4(s), an identical variation tendency is observed between the overlap population and the bond length, that is the longerbond length of C��M for Fe11CrC4(s) simultaneously corresponding to the larger overlap population. For the specific behavior of C��M in Fe11CrC4, it should come from the diversity of bond angles induced by Cr atoms in different doping sites. Putting the anomaly aside, it��s acceptable that Cr dopant could enhance the covalent bond strength of C��M according to the overlap population per unit of bond length. In consideration of the surprisingly sensitivity of C-M bond for doped site, it can be sure that the covalent bond net is constituted by the covalent bond chains of M��C��M, existing in the Fe11CrC4 as well as Fe3C and Cr3C, just like the previous findings in Cr28-xFexC12 [27].
Table 3 Bond type, bond length and overlap population of Fe3C, Fe11CrC4, and Cr3C
In the description of Fig. 2, the electric spin status of each kind atom is shown and the remarkable asymmetry between up-spin and down-spin indicates an non-ignorable magnetic property except for Cr3C. For d orbitals of Fe in each kind of carbide and those of Cr in Fe11CrC4, the large spin splitting occurs near the Fermi level. Specifically, the up-spin channel of Fe-d orbital is nearly completely occupied and the down-spin channel is partially occupied, opposite to the case of Cr, thus leading to the contrary for the sign of Fe and Cr in magnetic moments (Ms) as given in Table 4. Furthermore, the magnetic moments of Fe(s) and Fe(g) in Fe3C are respectively 1.92 ��B/atom and 1.82 ��B/atom, which are slightly smaller in contrast to other DFT calculations.
Once Fe locating in the general or special site substituted by Cr, the magnetic moments of the rest of Fe atoms in cell all reveal an obvious reducing tendency comparing to those in Fe3C. While the difference value between Fe(s) and Fe(g) increases,0.16 ��B/atom, 0.24 ��B/atom respectively for Fe11CrC4(g) and Fe11CrC4(s). As for Cr atoms in the Fe11CrC4, Cr(g) possesses a larger magnetic moment with spin-down. On the other hand, the spin polarization of C atoms resulting from the hybridization with the neighboring metal atoms, the magnetic moment also declines when Fe atoms are replaced by Cr, of which C nearest to Cr atom in the doped structure has the minimum, -0.06 ��B/atom, and -0.05 ��B/atom for the ��general�� and ��special�� sites severally known from Mulliken population analysis. The magnetic moments per cell unit of Fe3C, Fe11CrC4(g) and Fe11CrC4(s) are 20.36 ��B, 16.92 ��B, and 17.62 ��B respectively. It isn��t beyond doubt that Cr-doped decreases the magnetic moments of Fe3C. Meanwhile, the variation for magnetic moments of Fe11CrC4 is related to doping sites. In other words, Cr doping in Wyckoff positions of 8d will cause a lower magnetic moment.
Table 4 Calculated spin magnetic moments of Fe3C and Fe11CrC4

3.3 Dielectric properties
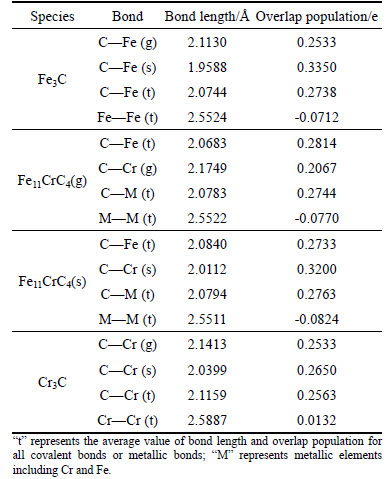
As the experiment shown, high-carbon ferrochrome powders (HCFCP) exhibit the weakly paramagnetic behavior as their permeability is slightly less than 1, and the value gradually decreases with microwave frequency rising [15]. Other than permeability, the permittivity of HCFCP could achieve a relatively high value. So, the heating process of HCFCP in the microwave filed is prevailingly determined by the dielectric properties, of which the main phase (Cr, Fe)7C3 makes a significant contribution to it. In the case of (Fe, Cr)3C, as a sort of the ferromagnetic phase, its permeability couldn��t be overlooked due to the high magnetic susceptibility. Considering the limit of calculation by CASTEP in permeability, the dielectric property is still a key point in current paper. For the mechanism of dielectric properties at optical frequencies, it��s acknowledged of electronic transitions from the occupied states to the unoccupied states through the interaction with energy quantum (namely photons in optical frequency) [28-29], and Fig. 4 displays the real part (��r') and imaginary part (��r") of the permittivity for Fe3C, Fe11CrC4(g), Fe11CrC4(s), and Cr3C as a function of frequency (v=E/h, where v, h, E represents frequency, planck constant and energy respectively). Generally speaking, the imaginary part, namely the relative dielectric loss factor, is calculated by the matrix elements between the occupied and unoccupied electronic states; while the real part, namely the relative permittivity, is derived from the imaginary part using the Kramers�CKronig relationship [30]. From Fig. 4, it is observed that the sequence of both ��r' and ��r" follows the order: Fe11CrC4(s)>Cr3C>Fe11CrC4(g)>Fe3C in the range of infrared frequencies (h��=1.24��10-3 eV to 0.5 eV and h��=1.24��10-3 eV to 1.2 eV respectively). Thus, it can be obtained that the permittivity of Fe11CrC4 is greater than that of Fe3C and concretely there exists an intimate correlation between permittivity and Cr-doped site. When exceeding 0.5 eV and 1.2 eV for ��r' and ��r",the former sequence will be broken. Previous studies [14, 31] have pointed out that the permittivity has the close relations with the covalent bond strength of C��M as well as the metallic anti-bonding. As Table 3 described, the covalent bond of C��M follows the order of Fe11CrC4(s)>Fe11CrC4(g)>Fe3C>Cr3C corresponding to the overlap population (e/) of 0.1328, 0.1320, 0.1319, 0.1211, basically in concert with the ranking of the permittivity apart from Cr3C at the low frequency as displayed in Fig. 4. As predicted, the sequence of the metallic anti-bonding is opposite to the order of the permittivity at high frequency. In conclusion, the contribution to the dielectric properties at the low frequency is mainly derived from the covalent bond, and beyond a certain frequency, the metallic anti-bonding will play the dominant role. Here, we offer the reasonable evidence for above interpretation. The wider hybridization area in DOS, equal to covalent bond strengthening, results in covalent electrons remarkable delocalization, meaning more effortless electron transition along with more effective interactions with energy quantum, which promotes the atomic thermal vibration. Since the 4s and 4p electrons of Fe(Cr) can form the anti-bonding orbitals and strongly reflect energy quantum, the inverse relation with permittivity can be explained. In particular, the stronger ionicity of Cr3C should take account for its specificity in dielectric performance.
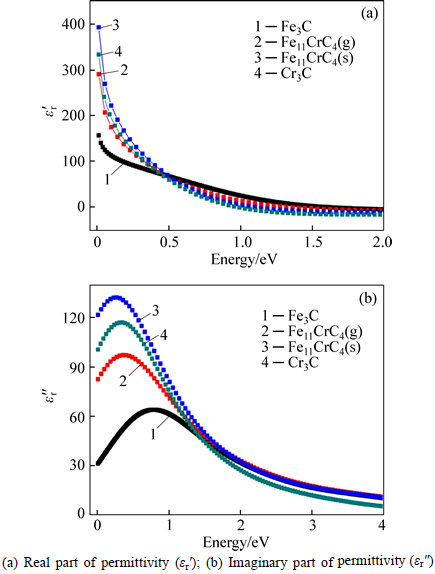
Fig. 4 Calculated dielectric function of Fe3C, Fe11CrC4, and Cr3C:
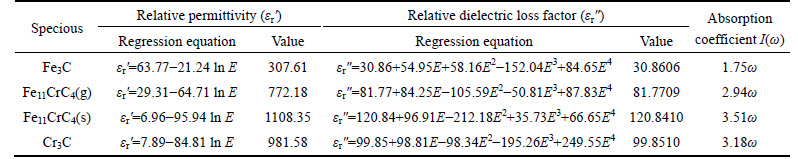
As Fig. 2 shown, 3d electrons of Fe(Cr) not only form covalent bonds with 2p electrons of C, but also form metallic bonds, thus being weak bound electrons. Meanwhile no band gap between the valence band and conduction band, the inter-band transition of 3d electrons prefers to occur in microwave band even if not obtaining high energy. Based on above evidences, the dielectric properties at microwave frequencies (h��=1.24��10-6 eV to 1.24��10-3 eV) can still be indirectly calculated in terms of the non-linear regression of the dielectric properties at infrared frequencies in spite of first- principles calculations of the dielectric properties limited to optical frequencies, and this method also has been applied to the calculation of permittivity for Fe-doped Cr7C3 and Mn7C3 [14, 31]. The detailed expressions accompanied the corresponding numerical values when microwave frequency is 2.45 GHz (h��=1.02��10-5 eV) for heating are shown in Table 5, in addition to the absorption coefficient I(��) calculated by [28]:
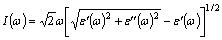 (2)
(2)
where �� represents the angular frequency of the microwave field.
Exactly as Table 5 depicted, when microwave frequency is 2.45 GHz, the sequences of both ��r' and ��r" for Fe3C, Fe11CrC4, and Cr3C are consistence with the infrared dielectric properties. Moreover, I(��) reveals the same order as ��r' and ��r", signifying the best temperature rising characteristic of Fe11CrC4(s). In consideration of the same large magnitudes of Fe-doped Cr7C3 as Cr-doped Fe3C for the permittivity in the microwave band [14], it seems unimaginable of the extremely low permittivity of HCFCP [15]. Besides the inevitable deviation between the theoretical calculation and actual measurement, the major reason is attributed to the powdering of massive material. The energy gap of the material increases with the size of the crystals decreasing [32], resulting in the increment of the localization of molecular orbitals and thus making electron transition difficult, of course causing the permittivity consequentially dropping in the extent of microwave frequency. Nevertheless, the occurrence of red shift of the dielectric properties with particle size reducing can prevent ��r" from sharply lowering to some degree in the microwave band [31].
Besides the contribution of electron transition, the dielectric properties of above carbides are also influenced by the polarization of electrons or atoms in the microwave electromagnetic field, which is analogous to dipole polarization. Under the electrical field force, nearly each atom would move along with the electric field direction and deviate from orginal lattice sites, generating polarization. Owing to the large gap in atomic radius between C and Cr (Fe), C atoms may jump through the lattice clearance once getting enough energy, thus constituting defect dipole with the metal ions or anion vacancies. Since the strength of covalent bonds in M-C-M chains is far weaker than that of common ones, the freedom of the relevant atoms is remarkable, preferring to demonstrate more significant polarization.
In terms of the undulatory theory, a reasonable interpretation also can be given for the changing of the permittivity in Fig. 4. As generally acceptable, the longer wavelength corresponds to the better penetrability in materials, namely energy quantum with lower energy can reach the depth of carbides followed the enlargement of the effective impact with valence electrons, meaning the higher permittivity. Even if the first principle calculation can��t precisely characterize the real case of these compounds with the limitation of DFT, it still offers a theoretical guidance for the future research and practical production in the fields of materials and metallurgy.
Table 5 Nonlinear regression equation of permittivity of Fe3C, Fe11CrC4 and Cr3C (E stands for energy)
4 Conclusions
In summary, a completely theoretical calculation has been carried out to analyze the electronic structure, magnetic and dielectric properties of Cr-doped Fe3C as well as Fe3C and Cr3C. The enlargement of formation enthalpy of Fe11CrC4 mainly comes from the stronger metal anti-bonding, leading to the lowering of stability. The more thermal excited electrons should be responsible for the stronger metallicity in dopant. Similarly, the more shared electrons offer a reinforce for the covalent bonds of C-M, even though the bond length has an increasement due to the existence of Cr. The calculation values for magnetic moments of unit cell are in good agreement with previous theoretical results. The Cr-doped Fe3C has a remarkable influence on the local magnetic moments for other atoms in unit, indicating the strong interaction among atoms. For either Fe11CrC4(g) or Fe11CrC4(s), the local magnetic moment of Fe at 4c site is larger than that at 8d site in contrary to Cr. The stronger covalent bond of Cr-doped Fe3C originating from the improving electron delocalizability makes the inter-band transition more easily, implying the higher permittivity. The effect of metallic anti-bonding gradually plays a leader as frequency rises; meanwhile the penetrativity of energy quantum increases with the energy lowering.
References
[1] ZHOU L, LIU G, MA X L, LU K. Strain-induced refinement in a steel with spheroidal cementite subjected to surface mechanical attrition treatment [J]. Acta Mater, 2008, 56(1): 78-87.
[2] KIM B, CELADA C, SOURMAIL T. The effect of silicon on the nanoprecipitation of cementite [J]. Acta Mater, 2013, 61(18): 6983-6992.
[3] CHAO J, SRINIVASAN S G. Unexpected strain-stiffening in crystalline solids [J]. Nature, 2013, 496: 339-342.
[4] HALLSTEDT B, DJURVOIC D, APPEN J, DICK A. Thermodynamic properties of cementite (Fe3C) [J]. Calphad, 2010, 34: 129-133.
[5] LV Z Q, ZHANG F C, SUN S H, WANG Z H, JIANG P, ZHANG W H, FU W T. First-principles study on the mechanical, electronic and magnetic properties of Fe3C [J]. Comput Mater Sci, 2008, 44(2): 690-694.
[6] LV Z Q, FU W T, SUN S H, BAI X H, GAO Y, WANG Z H, JIANG P. First-principles study on the electronic structure, magnetic properties and phase stability of alloyed cementite with Cr or Mn [J]. J Magn Magn Mater, 2011, 323(7): 915-919.
[7] GAO Yang, WANG Bo, GUO Ming-wei, LV Zhi-qing, SUN Shu-hua, ZHANG Rong-hua, FU Wan-tang. First-principles study on surface structural, magnetic and electronic properties of alloyed cementite with Co or Ni [J]. Comput Mater Sci, 2014, 85: 154-158.
[8] NIKOLUSSI M, SHANG S L, GRESSMANN T, LEINEWEBER A, MITTEMEIJER E J, WANG Y, LIU Z K. Extreme elastic anisotropy of cementite, Fe3C: First-principles calculations and experimental evidence [J]. Scripta Mater, 2008, 59(8): 814-817.
[9] GARVIK N, CARREZ P, CORDIER P. First-principles study of the ideal strength of Fe3C cementite [J]. Mater Sci Eng A, 2013, 572: 25-29.
[10] GAO Yang, LV Zhi-qing, SUN Shu-hua, QU Ming-gui, SHI Zhong-ping, ZHANG Rong-hua, FU Wan-tang. First principles study on surface structure and stability of alloyed cementite doped with Cr [J]. Mater Lett, 2013, 100: 170-172.
[11] ZHOU C T, XIAO B, FENG J, XING J D, XIE X J, CHEN Y H, ZHOU R. First principles study on the elastic properties and electronic structures of (Fe, Cr)3C [J]. Comput Mater Sci, 2009, 45(4): 986-992.
[12] IGNATENKO M, TANAKA M. Effective permittivity and permeability of coated metal powders at microwave frequency [J]. Phys B, 2010, 405(1): 352-358.
[13] HAO Jiu-jiu, CHEN Jin, HAN Pei-de, LI Kai, SUN Hong-fei. Solid phase decarburization kinetics of high-carbon ferrochrome powders in the microwave field [J]. J Iron Steel Res Int, 2014, 85(3): 461-465.
[14] CHEN Zhi-liang, CHEN Jin, HAN Pei-de, HAO Jiu-jiu, LIN Wan-ming. First-principles calculation of electronic structure and microwave dielectric properties of Fe-doped o-Cr7C3 [J]. Comput Mater Sci, 2014, 83: 298-302.
[15] LI Wei, CHEN Jin, GUO Li-na, HAO Jiu-jiu, HAN Pei-de, LIU Jin-ying. Electromagnetic properties of high-carbon ferrochrome powders decarburized in solid phase by microwave heating [J]. Mater Sci Eng B, 2014, 189: 58-63.
[16] FROIDEVAL A, IGLESICS R, SAMARAS M, SCHUPPLER S, NAGEL P, GROLIMUND D. Magnetic and structural properties of FeCr alloys [J]. Phys Rev Lett, 2007, 99: 237201.
[17] WU Z G, COHEN R E. More accurate generalized gradient approximation for solids [J]. Phys Rev B, 2006, 73: 235116.
[18] PERDEW J P, BURKE K, WANG Y. Generalized gradient approximation for the exchange-correlation hole of a many-electron system [J]. Phys Rev B, 1996, 54: 16533-16539.
[19] CORSO A D. Density-functional perturbation theory with ultrasoft pseudopotentials [J]. Phys Rev B, 2001, 64: 235118.
[20] TYUTEREV V G, VAST N. Murnaghan��s equation of state for the electronic ground state energy [J]. Comput Mater Sci, 2006, 38(2): 350-353.
[21] JIANG C, SRINIVASAN S G, CARO A, MALOY S A. Structural, elastic, and electronic properties of Fe3C from first principles [J]. J Appl Phys, 2008, 103: 043502.
[22] LIYANAGE L S, KIM S G, HOUZE J, TSCHOPP M A, BASKES M I. Structural, elastic, and thermal properties of cementite (Fe3C) calculated using a modified embedded atom method [J]. Phys Rev B, 2014, 89: 094102.
[23] SHEIN I R, MEDVEDEVA N I, IVANOVSKII A L. Electronic and structural properties of cementite-type M3X (M=Fe, Co, Ni; X=C or B) by first principles calculations [J]. Phys B, 2006, 371(1): 126-132.
[24] WUN C, CARTER E A. Structure and stability of Fe3C-cementite surfaces from first principles [J]. Surf Sci, 2003, 530(1/2): 87-100.
[25] LI Ye-fei, GAO Yi-min, XIAO Bing, MIN Ting, YANG Ying, MA Sheng-qiang, YI Da-wei. The electronic, mechanical properties and theoretical hardness of chromium carbides by first-principles calculations [J]. J Alloys Compd, 2011, 509(17): 5242-5249.
[26] ZHANG Yong-fan, LI Jun-qian, ZHOU Li-xin, XIANG Sheng-chang. A theoretical study on the chemical bonding of 3d-transition-metal carbides [J]. Solid State Commun, 2002, 121(8): 411-416.
[27] MUSIC D, KREISSIG U, MERTENS R, SCHNEIDER J M. Electronic structure and mechanical properties of Cr7C3 [J]. Phys Lett A, 2004, 326(5/6): 473-476.
[28] SAHA S, SINHA T P. Electronic structure, chemical bonding, and optical properties of paraelectric BaTiO3 [J]. Phys Rev B, 2000, 62: 8828-8834.
[29] HUANG Yu-hong, ZHANG Zong-quan, MA Fei, CHU P K, DONG Cui-ping, WEI Xiu-mei. First-principles calculation of the band structure, electronic states, and optical properties of Cr-doped ZnS double-wall nanotubes [J]. Comput Mater Sci, 2015, 101: 1-7.
[30] AKYURTLU A, KUSSOW A G. Relationship between the Kramers-Kronig relations and negative index of refraction [J]. Phys Rev A, 2010, 82: 055802.
[31] LI Wei, CHEN Jin, HAO Jiu-jiu, GUO Li-na, HAN Pei-de, CHEN Zhi-liang. Effect of Fe doping on electronic structure, chemical bonds and dielectric properties of o-Mn7C3 [J]. Phys Lett A, 2015, 379(18/19): 1219-1223.
[32] BEHESHTIAN J, SADEGHI A, AMAL M N, MICHEL K H, PEETERS F M. Induced polarization and electronic properties of carbon-doped boron nitride nanoribbons [J]. Phys Rev B, 2012, 86: 195433.
(Edited by YANG Hua)
Foundation item: Project(51174252) supported by the Joint Funds of the National Natural Science Foundation of China
Received date: 2015-08-24; Accepted date: 2015-12-27
Corresponding author: CHEN Jin, Professor, PhD; Tel: +86-13111009379; E-mail: chenjin2013815@126.com
Abstract: The first-principles calculations were performed to investigate the electronic structure, magnetic and dielectric properties of Cr-doped Fe3C, in comparison to those of pure Fe3C and Cr3C. The obtained results show that the thermodynamic stability of Cr- doped Fe3C becomes weaker in terms of the larger formation enthalpy, on the contrary, the metallicity and covalency are found to strengthen to some extent. The magnetic moments of Fe3C, Fe11CrC4(g), and Fe11CrC4(s) are respectively 21.36 ��B/cell, 16.92 ��B/cell, and 17.62 ��B/cell, and in Fe11CrC4(g) and Fe11CrC4(s), the Fe of Wyckoff positions of 8d and 4c is substituted by Cr. The local magnetic moment of Cr at 8d site is larger than that at 4c site in the doped structure, which is opposite to that of Fe. In low frequency band, the permittivity follows the ranking of Fe11CrC4(s)>Cr3C>Fe11CrC4(g)>Fe3C. Once exceeding a certain frequency, the sequence will be broken. Besides the electron transition, the polarization of atoms also makes a contribution to the dielectric properties.
