
���±�ţ�1004-0609(2011)04-0919-08
������(100)�������ʵ��ܶȷ������ۼ��㼰��Ը�ѡ��Ӱ��
������1���½���2, 3���� ��2���� ��3
(1. ������ѧ ��ѧ����ѧԺ������ 530004��
2. ������ѧ ��Դ��ұ��ѧԺ������ 530004��
3. ������ѧ ������ѧ�빤�̼���ѧԺ������ 530004)
ժ Ҫ�������ܶȷ�������(DFT)ƽ�沨���Ʒ����������������(100)����Ľṹ��ԥ��ԭ�ӵ�Mulliken�����Լ����ӽṹ�������ͻ����������е�ɷֲ��쳣��ԭ�Ӹ�ѡ�Ƕȷ�������ṹ�����ʶԻ�����ѡ��Ϊ��Ӱ�졣���������������(100)�����ԥ��С������Fe-S����������������ǿ������5��λ����ԭ�Ӿ��нϸߵĻ��ԣ����������ԭ�ӵ���϶���ͣ������ĵ�����ǿ������ģ�����ĵ绯ѧ������ǿ��
�ؼ��ʣ�
������������ܶȷ������ۼ�����Mulliken���������ӽṹ����ѡ��
��ͼ����ţ�TD923 ���� ���ױ�־�룺A
Density functional theory calculation of surface properties of pyrite (100) with implications for flotation
LI Yu-qiong1, CHEN Jian-hua2, 3, CHEN Ye2, GUO Jin3
(1. School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China;
2. School of Resources and Metallurgy, Guangxi University, Nanning 530004, China;
3. School of Physics Science and Engineering, Guangxi University, Nanning 530004, China)
Abstract: The structural relaxation, atomic Mulliken populations and electronic structures of ideal pyrite (100) surface were calculated using density functional theory (DFT) method of plane-wave pseudopotential method. The reason for the charge unconventionality in bulk pyrite was explained. Moreover, the effects of structures and properties of surface on pyrite flotation behavior were analyzed from a flotation point of view. The calculated results show that the relaxation of pyrite (100) surface is relatively small, and the Fe-S interaction increases at the surface compared to that in the bulk. The calculated electronic structure results suggest that the surface 5-coordinated Fe atom has high activity. The energy gap of surface Fe and S atoms decreases, suggesting higher conductivity, as well as higher electrochemical activity, of the surface layer than the bulk.
Key words: pyrite surface; density functional theory calculation; Mulliken populations; electronic structures; flotation
������㷺�����ڸ��ֿ��У���������ɫ����(��ͭ��Ǧ��п��������)�棬�ڲ�����ĭ��ѡ��������Щ��������ʱ�����������������ѡ��������⡣���⣬��ú����Ҳ�������ڻ��������ú�к��������꣬��ú��ѡ���У�ͨ�����ø�ѡ���ѳ���������ĸ�ѡ��Ҫ��ͨ��������ҩ����������ã���ˣ��о��������������ʶ����˽������ĸ�ѡ��Ϊ��ҩ�����û������зdz���Ҫ�����塣
Ŀǰ������������Ի���������ʵ������� ��[1-4]����������ļ���ģ����������뻷����������ֱ�ӵع۲쵽ԭ�Ӳ�ε���Ϣ�����������ǽ�һ���˽������������ʡ�HUNG��[5]����ƽ�沨���ƺ�˹���鷽�����������(100)��Ľṹ��ԥ�͵��ӽṹ��von OERTZEN��[6]����ab��ͷ�����ӻ�ѧģ��Ԥ�������(100)���������֮��IJ��죻CAI��PHILPOTT[7]����ab��ͷ�����Ʒ������������(100)�ı���̬�ͱ����ܡ�������ı���ṹ�����ʶԻ�����ѡ������Ҫ���壬Ŀǰ��������о���Ҫ�Ǽ����ڶԻ�����������ʵ����۷������棬�����ڻ����������������ɸ��ԵĹ�ϵ����ټ���������
�Ѿ��н϶�IJ����ܶȷ������۷���(DFT)��Ի�����������о��������[8-12]����������[13]�о��˻��������ࡣ����Щ������������Ļ������������ʣ��羧�����ȣ���ʵ��ֵ���ϵ÷dz��ã���������һЩ����û�еõ��ܺý������������������ԭ�Ӻ���ԭ�ӵ�Mulliken��ɲ��Ӽ������⣬Ŀǰ�����������ģ��Ľ����������[14]�⣬���������Ľ����������������������[6, 15]��������Ҳû�ж������Ľ����������Ľ��͡���������[16]����Ϊ����������3d��������γ���λ�����õ��϶����ԭ�ӶԵĵ��ӵ�Ե�ʣ���������縺��������ì��(��ĵ縺�Դ�����)�������������������磬�⽫������ѡ������ҩ����������������ӵ����ã�����ʵ���ϻ����������ļ�̬�����ġ�
��Ȼ��������Ļ�����Ϊ�������壬(100)����������Ҫ�Ľ����棬���������п��ܻ���S��S�����ѣ�Fe��S�����ѣ��������߶����ѣ����²�ͬ�ı����ࡣ�������ߵļ�����õ�����Fe��S���ѡ���������������S������(S2)������(100)���棬��������б���ṹ��ԥ��Mulliken���Ӽ����ӽṹ���㣬���۱���������֮��IJ��죬�����ݱ�����ӽṹ�����ʣ��о����������ҩ�����������õĵ绯ѧģ�ͺͻ�����
1 ���㷽��
����CASTEP����[17-18]���ܶȷ������ۿ��[19-22]�¶Ի�����(100)�������̬�ܶȡ��ܴ��ṹ�Ͳ��ӽ��е�һ��ԭ��ƽ�沨���Ƽ��㡣����ģ�ʹ��Ż����������г��������нṹ�Ż����ٽ������ʼ��㡣��ǰ���������м���Ļ�����[13]�����о��жԱ�������Ż������������Ϊ������(1��1)���浥��ģ�ͣ�����GGA�µ�PW91������������������270 eVƽ�沨�ض��ܺ�4��4��1��Monkhorst-Pack k��ȡ���� ��[23-24]�����ó�������[25]�������۵��Ӻͺ˵�����á������Ż���������Ϊԭ��λ�Ƶ�������ֵΪ0.000 2 nm��ԭ�Ӽ���������������ֵΪ0.8 eV��nm��ԭ�Ӽ����Ӧ��������ֵΪ0.1 GPa����������ı��������ֵΪ2.0��10-5 eV��atom-1����Ǣ������������Ϊ2.0��10-6 eV��atom-1��ԭ�ӵ����Ƽ���ѡȡ�ļ۵��ӷֱ�ΪS 3s23p4��Fe 3d64s2�������ʵļ�������뼸���Ż���ͬ�IJ���������̬�ܶȵ�ʱ����õ�smearingֵΪ0.1 eV����������ʱ�������ԭ�Ӷ������˳�ԥ���Բ�ͬԭ�Ӳ����ղ��Ƚ��в��ԣ���ȷ�������IJ��ԭ����������ģ����ɺ������ǵij�ԥ�����еļ��㶼�����������������ڵ��ռ��н��С�
2 ���������
�������峣�������壬����Ŀռ�ԳƽṹΪ![]() ������ʽΪFeS2�����ڵ��ᾧϵ��ÿ����������4��FeS2���ӵ�Ԫ����ԭ�ӷֲ�������������6�����ļ�8�������ϣ�ÿ����ԭ����6�����ڵ���ԭ����λ���γɰ����幹�죬��ÿ����ԭ����3����ԭ�Ӻ�1����ԭ����λ���γ������幹�죬������ԭ��֮���γ�����״�ṹ�����������(
������ʽΪFeS2�����ڵ��ᾧϵ��ÿ����������4��FeS2���ӵ�Ԫ����ԭ�ӷֲ�������������6�����ļ�8�������ϣ�ÿ����ԭ����6�����ڵ���ԭ����λ���γɰ����幹�죬��ÿ����ԭ����3����ԭ�Ӻ�1����ԭ����λ���γ������幹�죬������ԭ��֮���γ�����״�ṹ�����������(![]() )��ʽ���ڣ�������
)��ʽ���ڣ�������![]() �������С���Fe��S�����ѵ�����(100)�����ϣ���ԭ����5����ԭ����λ�����������ԭ����2����ԭ�Ӽ�һ����ԭ����λ��
�������С���Fe��S�����ѵ�����(100)�����ϣ���ԭ����5����ԭ����λ�����������ԭ����2����ԭ�Ӽ�һ����ԭ����λ��
��1��2���зֱ�Ϊ��ͬԭ�Ӳ�������ͬ��ղ��ȶԱ�����Ӱ��Ľ�����ӱ�1���Կ��������溬��15��ԭ�Ӻ����ܵı仯�Ѿ���С���ӱ�2�Ľ�
��1 ��ͬԭ�Ӳ����ı�����
Tabel 1 Energy of surface containing different atomic layer numbers
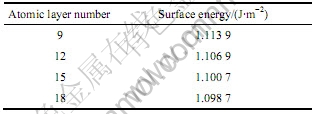
��2 ��ղ��ȶԱ����ܵ�Ӱ��
Tabel 2 Effect of vacuum layer thickness on surface energy

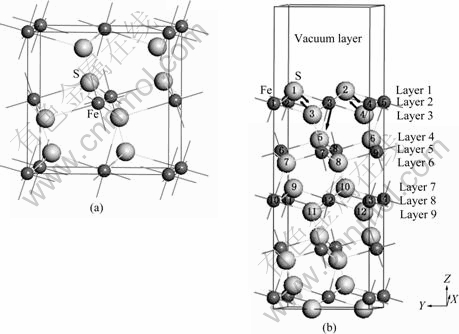
ͼ1 ���������(a)��������(100)���浥��(b)
Fig.1 Bulk pyrite unit cell (a) and pyrite (100) surface unit cell (b)
���ɿ�������ղ���Ϊ1.0 nm�ı���ı�������͡���ˣ�15��ԭ�Ӽ�1 nm����ղ��ȵı���ṹ�ܹ�����������������������������(100)���浥��ģ����ʾ��ͼ1�С�
������(100)����ṹ������λ��ʸ����ͼ1��ʾ����3����Ϊ��ԥ����漸��ԭ�ӵ���λ����λ�ơ����������������λ��Ϊ4��������λ��Ϊ6������Ļ�����(100)����ֲ���3����λ�����5����λ����������������������ɱ���ԭ����λ�����ͣ�����ԭ��ȱ����Χԭ�ӵ�������������˲�ͬ�̶ȵij�ԥ�����ͼ1�ͱ�3��֪�������һ��Sԭ��������ڲ���ԥ�������Եij�ԥ�ǵڶ���ı���Feԭ�ӣ����ڲ���ԥ�˴�Լ0.01 nm���������е�Sԭ��������ԥ������������������������(100)�澭���ij�ԥ�dz�С��û�в������Եı����ع����ã�����ROSSO��[2]��CHATURVEDI��[3]��ʵ����Խ��һ�£�Ҳ��HUNG��[5]�ļ�����һ�¡�ԭ�ӽ��ڶ���������������Եij�ԥ��������������ԭ�Ӿ�����С��λ�ƣ��������ھŲ�ԭ�ӵij�ԥ���Ժ��Բ��ƣ���˿�����Ϊ������1~3��Ϊ����㣬4~6��ԭ��Ϊ������㣬��7~9��ԭ���Ѿ���ȫ�������������ʣ�Ϊ����㡣
ͼ2��ʾΪ��������9��ԭ�ӵĵ�ɷֲ�����4����Ϊ��ԭ�Ӳ�ԭ�ӵ�Mulliken��ɲ��ӡ���ͼ2
�ɿ�������һ�����ԭ�Ӵ�����ɽϴ�(-0.11 e)����
��3 ԭ����λ��λ��
Table 3 Atomic coordination and displacements
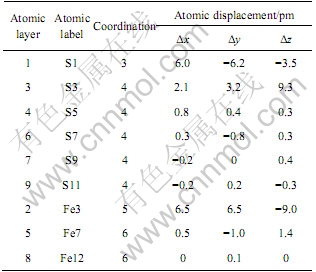
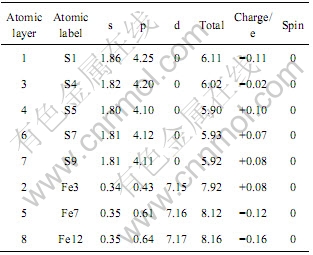
���������࣬��ԭ�������ĵ�ɾ�����һ���ɸ������Ĺ��̣�������ԭ�Ӵ����Ϊ+0.08 e(S9)��������ԭ�Ӵ������(+0.08 e)������������µĽ����༰������ԭ�Ӵ�����ɣ���������ԭ��(Fe12)������(-0.16 e)���ɱ�4�г��Ĺ�����ӿ�֪��������ԭ��(S1)��Ҫ��p����õ����ӣ�s����õ��������ӣ���S4
ԭ����Ҫ��p����õ����ӣ��ҵõ��ĵ��ӱ�S1ԭ�ӵ��٣����S1ԭ��ӵ�����ĸ����(-0.11 e)����S4ԭ�Ӵ����������(-0.02 e)��������ԭ����Ҫ��p���ʧȥ���Ӷ�������ɣ�d�����ʧȥ�������ӡ�

ͼ2 ����9��ԭ�ӵĵ�ɷֲ�
Fig.2 Atomic charge distribution of nine layer of surface
��4 ԭ�ӵ�Mulliken��ɲ���
Table 4 Mulliken charge populations of atoms
����ԭ�Ӻ���ԭ�ӵĵ��ӵ�ʧ�����֪���ڻ�����(100)�����ϣ��������ԭ�Ӻ���ԭ��֮�䷢����ת�ƣ�����ԭ����ת�Ƶ�����ԭ���ϣ�����NESBITT��[26]��Ի������������Ĺ۵�һ�£�������S��S�����ѵı����ϣ��ᷢ����ԭ��ʧȥ���ӱ���������ԭ�ӻ�õ��ӱ���ԭ�ķ�Ӧ����von OERTZEN��[6]����XPS�����Ի�����(100)������о�Ҳ���������ɴ���ԭ�ӵ���ԭ�ӵ�Ǩ�ơ����⣬ԭ������ֵ����(100)���ϵ�5��λ�������е���ԭ�Ӷ��������� �Եġ�
��ԭ�ӵ�Mulliken��ɲ��ӷ�����֪�������������е�����ԭ�������ĵ����ʵ�������෴��������ԭ��ȴ��ʵ�������ͨ���Ե��Ӳ���ܶȵķ��������Խ�һ���˽�������ֲ���Ŀ���ԭ����ǰ��Ի�����ṹ�Ľ��ܿ���֪�����ڻ����������У���ԭ������ԭ��֮��ɼ����Զ������(![]() )��ʽ���ڣ������������������ԭ��ֻ�����ԭ�ӳɼ�������ͬ���������ֲ�����������������������ԭ�ӵ�ɷֲ��ķ�����
)��ʽ���ڣ������������������ԭ��ֻ�����ԭ�ӳɼ�������ͬ���������ֲ�����������������������ԭ�ӵ�ɷֲ��ķ�����
ͼ3��ʾΪ����Fe��S��S��S��֮���Լ����� Fe��S��֮��ĵ�ɲ���ܶȡ�ͼ����ɫ�����������ȱʧ����ɫ����������Ӹ�������ɫ���������ܶȼ���û�з����仯������ͼ3(a)���Կ������������ڶ���ԭ��֮�主������������������ԭ��֮����Ӹ���������ǿ�ҵĵ����ų����ã���ƫ��������λ����ԭ�ӣ���������ԭ����Χ�ĵ���ȱʧ����ԭ�Ӵ�����ɡ�ͼ3(b)��������ʾ��������ԭ����Χ�������Ƹ��ǡ�ͼ3(c)��ʾ�˱���Fe��Sԭ��֮��ĵ��ӷֲ�״��������Fe3ԭ�ӿ�����ղ�һ������ȱ����һ����λ��ԭ�ӣ���������һ����ȱʧ�ģ���ԭ�Ӵ�����ɡ�
���⣬��ͼ3(c)�������Կ������ڱ���S1�ϲ������Եĵ����Ƹ���������Щ��������Fe3ԭ����ɢ�������������Ӵӱ�����ԭ������ԭ��ת�ƣ�����S1ԭ�Ӵ�����ɡ��ɴ˿��Կ���������ļ���������ʵ������ģ�ͬʱ��Ҳ�������ѡʵ��һ�£����ҩ�ڻ�������������ӵĻ�ѧ�������ã��Լ����������軯�����������������ӵ����õȡ����⣬���ڻ�����ĵ绯ѧ���ռ���ѡ��ֻ�б�����ԭ�Ӵ�����ɣ����ܺ������ͻ�����������Ӹ�����������۵�Ԫ����[27]��
Mulliken�����ص����ӿ����������Ƽ��Ĺ��ۺ������ԣ��ϸߵIJ���ֵ�������ʹ����ԣ����ϵ͵IJ���ֵ������֮������������[28-29]����5����Ϊ���Ӧ�ı��������ԭ�ӵļ��IJ��Ӻͼ���(S1��S2��S3��S4��S5�ֱ���S9��S10��S11��S12��S13��λ�����Ӧ)�������ļ��IJ���ֵ�������еļ��IJ���ֵ�������Բ��죬����Ҳ��ͬ���ɱ�5��֪������Fe3ԭ�����һ��S1��S2ԭ��֮��ļ��IJ���ֵ��������Fe12ԭ����S9��S10ԭ��֮����IJ���ֵ���������涥����-��ԭ��֮��Ĺ�������ǿ������Fe3ԭ���������S3��S4ԭ��֮��IJ���ֵС������Fe12ԭ����S11��S12֮��ļ��IJ���ֵ����������֮��Ĺ����Լ���������������ǿ��S5��Fe3��֮��IJ�
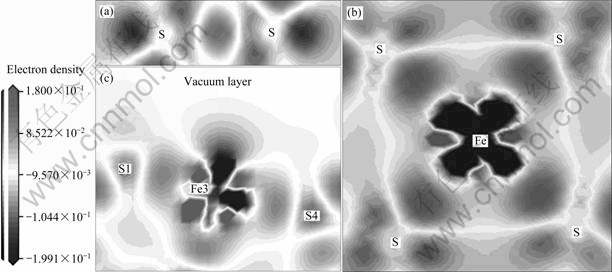
ͼ3 ��ɲ���ܶ�ͼ
Fig.3 Electron density difference map: (a) Two coordinated S atoms; (b) Bulk Fe and its four coordinated S atoms; (c) Surface Fe and its two coordinated S atoms
��5 ԭ�ӵ�Mulliken���IJ���
Table 5 Mulliken bond population of atom
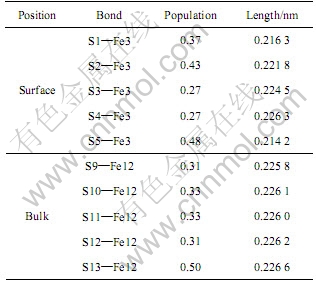
��ֵ��S13��Fe12�Ľӽ������Fe��Sԭ��֮��Ĺ�����ǿ���仯�����⣬��S4��Fe3��S12��Fe12֮��ļ����ӽ��⣬�����Fe��Sԭ��֮��ļ�����С����������Ӧԭ��֮��ļ���������������ϵ� Fe��Sԭ��֮�������ñ������еĸ�ǿ��
�����ϵ�ԭ��(Fe3��S1��S4)�������Ļ����������е�ԭ�Ӳ�ͬ������ͨ���Ƚ�����������༰����ԭ��(F37��S5��S7)��̬�ܶȣ�����������˽������ӽṹ�ı仯��ͼ4��5�ֱ���ʾ�˲�ͬԭ�Ӳ����ԭ�Ӻ���ԭ�ӵ�̬�ܶȡ���ͼ4��֪(Fe 4s����Ĺ��dz�С��δ��ͼ����ʾ)��������Ƚϣ�������ԭ��(Fe3)��3d̬��-2 eV����̬�ܶȷ�ǿ������ǿ����ͼ5����ԭ��̬�ܶ�Ҳ��ʾ����������ԭ����ȣ�����S1ԭ�ӵ�3p̬��-2.5~0 eV֮����ܴ��Ĺ���������ǿ����-2 eV����̬�ܶȷ�ǿ������ǿ��Fe��Sԭ�ӵ�̬�ܶ���������ص�������Fe��S֮����������ǿ������Fe��Sԭ����0.8 eV���������Ҳ��ǿ�ˡ�S5��S7��S9ԭ�ӵ�̬�ܶȽ�Ϊ���ơ�
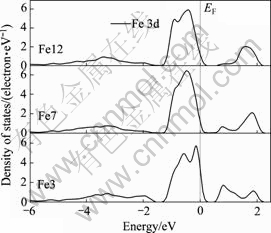
ͼ4 ��ԭ��̬�ܶ�
Fig.4 Density of states of Fe atom
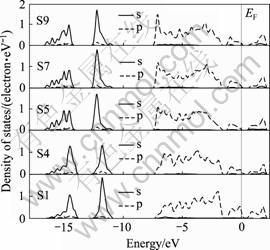
ͼ5 ��ԭ��̬�ܶ�
Fig.5 Density of states of S atoms
�ڸ�ѡʵ���У����������������ܵ����Ƶ�����ر����ڼ��Խ����У�������ɸ�������������������ԭ�ӵĻ����йء���ͼ4�ɼ��������������������(Fe3)3d̬��Ҫ�ڷ����ܼ�����������������������������ԭ�ӵĻ��Ժܴ��Ӹ����������������ԭ�ӷ�������������ڼ��Խ����л�������������������(���Ӹ�����)�������ã��ڱ����γ������������Ӷ������������á�
��һ���棬��ͼ4��5���Կ�����������Ƚϣ�������ԭ��(Fe3)����ԭ��(S1)����϶�����ˣ���ԭ��̬�ܶ����Դ��������ܼ�(����ԭ�ӵ�̬�ܶ������������ܼ���������Ϊsmearing���ȵ�ȡֵ��ԭ��)����˻�����(100)�������һЩ�ƽ��������������������Ʒ���й�ѧ�����������������һ�£�Ҳ��HUNG��[5]�ļ�����һ�¡�
��������һ�ְ뵼�������ĸ�ѡ�����漰�绯ѧ��Ӧ[30]����ͳ�ĵ绯ѧ���۸��ݻ�����λ�����ͻ�ҩ�ڻ������������������ڻ�ҩ������˫��ҩ��ƽ���λС�ڻ�����ľ���λ��ʱ�������Ϊ˫��ҩ����֮������������˫��ҩ[31]������λ���۲��������������ڻ���������ת�������ͬʱҲ���ܲ������ڻ������������ã��������ڻ���������ת�ƺ����������ǻ�����ѡ�绯ѧ���̵���Ҫ���ء�ǰ��ļ�����������������������������������ĵ�������ǿ������ĵ绯ѧ����Ҳ��˸�ǿ�����ڻ�����������н����������������а뵼��������һ����ԣ��뵼��ķ����ܼ��Ƚ�������Ҫ�ͣ���ˣ������дӻ���������������ת�Ƶ����ƣ��Ӷ����»����������е�����������������ҩ�����ڻ���������ҩ�ĵ��ӽ�������������ת�ƣ��������ഫ�ݣ�������˫��ҩ������������е��������ʵ������ϡ������������ֻ���ڻ�ҩ��ʱ���ڵ��Ӵӱ���������ת�ƹ����л��γ�Ф�ػ����ݣ���ֹ���ӵĴ��ݣ��Ӷ��谭��ҩ�Ľ�һ����������ˣ������������»�������������γɽ϶��˫��ҩ�������ڻ�����ĸ�ѡ����������������������������Ե��ӵ�������Զ���ڻ�����ģ���˻��������ĵ��ӻ�����ת�ƣ�������������ת�ƣ������ֻ����£����γ��˻��������ҩ��Ӧ�ĵ绯ѧ�������[32]������ҩ�ڻ��������ʧȥ���ӷ���������Ӧ�γ�˫��ҩ������������ӻ��������õ����ӷ���������Ӧ���Ӷ������ڻ�����ĸ�ѡ��
3 ����
1) ������(100)�澭����С�ij�ԥ���ҳ�ԥ��Ҫ�����ڱ�����λ�������ԭ����Χ������δ�������Ե��ع�����
2) ��ԭ�ӵĵ�ɾ�����һ���ɱ��浽������������Ĺ��̣�����ԭ�������˴Ӹ������Ĺ��̣������ڱ�����ԭ�Ӻ���ԭ��֮��ת�ƣ��ɱ�����ԭ��ת�Ƶ�������ԭ���ϡ�
3) ������-��ԭ��֮��������ǿ�������е���-��ԭ�ӡ�
4) ��������λ����ԭ�����������Եģ�������ԭ�ӵĻ��Ժܴ�����ڼ��Խ����л�������������������(���Ӹ�����)�������ã��ڱ����γ������������Ӷ������������á�
5) ������(100)��������϶�����ˣ������������������ǿ���������һЩ�ƽ�������������ĵ绯ѧ����Ҳ��ǿ�ˣ������дӻ���������������ת�Ƶ����ƣ�������Щ�������Ļ����Ͻ����˻�ҩ�������������������õĵ绯ѧģ�͡�
REFERENCES
[1] LAAJALEHTO K, KARTIO I, SUONINEN E. XPS and SR-XPS techniques applied to sulphide mineral surfaces[J]. International Journal of Mineral Processing, 1997, 51(1): 163-170.
[2] ROSSO K M, BECKER U, FOCHELLA M F. Atomically resolved electronic structure of pyrite {100} surfaces: An experimental and theoretical investigation with implications for reactivity[J]. American Mineralogist, 1999, 84(10): 1353-1548.
[3] CHATURVEDI S, KATZ R, GUEVREMONT J, SCHOONEN M A A, STRONGIN D R. XPS and LEED study of a single-crystal surface of pyrite[J]. American Mineralogist, 1996, 81(1/2): 261-264.
[4] LEIRO J A, MATTILA S S, LAAJALEHTO K. XPS study of the sulphur 2p spectra of pyrite[J]. Surface Science, 2003, 547(1): 157-161.
[5] HUNG A, MUSCAT J, YAROVSDY I, RUSSO S P. Density-functional theory studies of pyrite FeS2 (100) and (110) surfaces[J]. Surface Science, 2002, 513(3): 511-524.
[6] von OERTZEN G U, SKINNER W M, NESBITT H W. Ab initio and XPS studies of pyrite (100) surface states[J]. Radiation Physics and Chemistry, 2006, 75(11): 1855-1860.
[7] CAI J, PHILPOTT M R. Electronic structure of bulk and (001) surface layers of pyrite FeS2[J]. Computational Materials Science, 2004, 30(3/4): 358-363.
[8] EDELBRO R, SANDSTR ? M ?, PAUL J. Full potential calculations on the electron bandstructures of sphalerite, pyrite and chalcopyrite[J]. Applied Surface Science, 2003, 206(1/4): 300-313.
[9] von OERTZEN G U, JONES R T, GERSON A R. Electronic and optical properties of Fe, Zn and Pb sulfides[J]. Physics and Chemistry of Minerals, 2005, 32(4): 255-268.
[10] WONES M, KARNATAK R C, ESTEVA J M, LEFEBVRE I, ALLA G, OLIVIER-FOURCADE J, JUMAS J C. Electronic structures of FeS and FeS2: X-ray absorption spectroscopy and band structure calculations[J]. Journal of Physics and Chemistry of Solids, 1997, 58(2): 345-352.
[11] von OERTZEN G U, SKINNER W M, NESBITT H W. Ab initio and x-ray photoemission spectroscopy study of the bulk and surface electronic structure of pyrite (100) with implications for reactivity[J]. Physical Review B, 2005, 72(23): 235427-1- 235427-10.
[12] OPAHLE I, KOEPERNIK K, ESCHRIG H. Full potential band structure calculation of iron pyrite[J]. Computational Materials Science, 2000, 17(2/4): 206-210.
[13] ������, �½���, �� ��. ��λȱ�ݻ�����ĵ��ӽṹ���両ѡ��Ϊ[J]. ������ѧѧ��, 2010, 26(5): 1435-1441.
LI Yu-qiong, CHEN Jian-hua, CHEN Ye. Electronic structures and flotation behavior of pyrite containing vacancy defects[J]. Acta Physico-Chimica Sinica, 2010, 26(5): 1435-1441.
[14] HUNG A, MUSCAT J, YAROVSKY I, RUSSO S P. Density-functional theory studies of pyrite FeS2 (111) and (210) surfaces[J]. Surface Science, 2002, 520(1): 111-119.
[15] von OERTZEN G U, SKINNER W M, NESBITT H W, PRATT A R, BUCKLEY A N. Cu adsorption on pyrite (100): Ab initio and spectroscopic studies[J]. Surface Science, 2007, 601(24): 5794-5799.
[16] ������, ������, ��ˮ��. ��������븡ѡ���������ӻ�ѧ�о�[J]. �й���ɫ����ѧ��, 1991, 1(1): 15-23.
WANG Dian-zuo, LONG Xiang-yun, SUN Shui-yu. Quantum chemical study on the mechanism of oxidation and flotation of sulfide minerals[J]. The Chinese Journal of Nonferrous Metals, 1991, 1(1): 15-23.
[17] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROVERT M I J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Zeitschrift fuer Kristallograhie, 2005, 220(5/6): 567-570.
[18] SEGALL M D, LINDAN P J D, PRONBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: ideas, illustrations and the CASTEP code[J]. Journal of Physics: Condensed Matter, 2002, 14(11): 2717-2744.
[19] лϣ��, ½ ��. �����ܴ�����[M]. �Ϻ�: ������ѧ������, 1998: 1-26.
XIE Xi-de, LU Dong. Energy band theory of solids[M]. Shanghai: Fudan University Press, 1998: 1-26.
[20] MARZARI N, VANDERBILT D, PAYNE M C. Ensemble density-functional theory for ab-initio molecular dynamics of metals and finite-temperature insulators[J]. Physical Review Letters, 1997, 79(7): 1337-1340.
[21] JONES R O, GUNNARSSON O. The density functional formalism, its applications and prospects[J]. Reviews of Modern Physics, 1989, 61(3): 689-746.
[22] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review, 1965, 140(4A): A1133-A1138.
[23] MONKHORST J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192.
[24] PCAK J D, MONKHORST H J. Special point for Brillouin-zone integrations��A reply[J]. Physical Review B, 1977, 16(4): 1748-1749.
[25] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990, 41(11): 7892-7895.
[26] NESBITT H W, BANCROFT G M, PRATT A R, SCAINI M J. Sulfur and iron surface states on fractured pyrite surfaces[J]. American Mineralogist, 1998, 83(9/10): 1067-1076.
[27] ��÷��, �½���, ���ټ�, ������. ������绯ѧ��ѡ�о�[J]. ������ѧѧ��: ��Ȼ��ѧ��, 2002, 27(4): 281-283.
YANG Mei-jin, CHEN Jian-hua, MA Shao-jian, HU Zhi-liu. Research of electrochemical flotation of pyrite[J]. Journal of Guangxi University: Natural Science Edition, 2002, 27(4): 281-283.
[28] SEGALL M D, SHAH R, PICKARD C J, PAYNE M C. Population analysis in plane wave electronic structure calculations[J]. Molecular Physics, 1996, 89(2): 571-577.
[29] SEGALL M D, SHAH R, PICKARD C J, PAYNE M C. Population analysis of plane-wave electronic structure calculations of bulk materials[J]. Physical Review B, 1996, 54(23): 16317-16320.
[30] �½���, ������, ¬����. �绯ѧ���ظ�ѡ�ܴ�ģ�ͼ�Ӧ��(��)����ҩ���������õ��ܴ�ģ��[J]. �й���ɫ����ѧ��, 2000, 10(3): 426-429.
CHEN Jian-hua, FENG Qi-ming, LU Yi-ping. Energy band model of electrochemical flotation and its application (��)��Energy band model of xanthate interacting with sulphide minerals[J]. The Chinese Journall of Nonferrous Metals, 2000, 10(3): 426-429.
[31] ALLISON S A, GOOLD L A, NICOL M J, GRANVILLE A. A determination of the products of reaction between various sulfide minerals and aqueous xanthate solution, and a correlation of the products with electrode rest potentials[J]. Metallurgical and Materials Transactions B, 1972, 3(10): 2613-2618.
[32] ������, �� ݣ. ���︡ѡ�绯ѧ[M]. ��ɳ: ���Ϲ�ҵ��ѧ������, 1992: 70-79.
FENG Qi-ming, CHEN Jin. Electrochemistry of sulfide mineral flotation[M]. Changsha: Central South University of Technology Press, 1992: 70-79.
������Ŀ��������Ȼ��ѧ����������Ŀ(50864001)
�ո����ڣ�2010-04-15�������ڣ�2010-12-28
ͨ�����ߣ��½��������ڣ���ʿ���绰��0773-3232200��E-mail: jhchen1971@sina.com
