
���±�ţ�1004-0609(2007)03-0465-06
�������������»�ͭ��ĵ绯ѧ��Ϊ
¬��������С�ԣ���������ŷ�������Ź���
(���ϴ�ѧ ��Դ�ӹ������﹤��ѧԺ����ɳ 410083)
ժ��Ҫ�����¶�Ϊ25 �漰pH=2�������£�ͨ��ѭ���������ͺ��λI��t�����о��˻�ͭ������ĵ绯ѧ�ֽ���Ϊ��ͨ��ѭ���������߷��֣���λ��400~800 mV(vs SHE)��Χ�ڣ���ͭ��缫���������������Ӧ������С����Ҫ���������ɵ��м������ѱ���һ�������ֽ⣬�Ӷ������˶ۻ�������λС��-400 mV (vs SHE)ʱ����ͭ��������ԭ��Ӧ�����ϴ����е�Fe3+�ܽϿ���ܽ�������������м����(ͭ������)��������λ�·�����ǿ�����������ֽⷴӦ���������Ӧ��һ�����ۻ�����ͭ���������ԭ��Ӧ��ǿ�ң��ҶԻ�ͭ����������������Ҫ���塣���⣬���λI��t����Ҳ֤ʵ�����Ͻ��ۡ�
�ؼ��ʣ�
��ͭ�����ֽ�������������������ԭ��
��ͼ����ţ�TD 952���� ���ױ�ʶ�룺A
Electrochemical behavior of chalcopyrite at normal temperature in acidic solution
LU Yi-ping, JIANG Xiao-hui, FENG Qi-ming, OU Le-ming, ZHANG Guo-fan
(School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China)
Abstract: The electrochemical reaction behaviors on chalcopyrite were investigated using cycle voltammetry and potentiostatic I��t curve at 20 �� and pH=2. The voltammograms show that the oxidization of chalcopyrite occurs at slow rate in potential range from 400 to 800 mV(vs SHE). The reason is that the intermediate products are very difficult to be further dissolved unceasingly if Fe3+ ion in lattice has not been dissolved. Fe3+ ion in lattice can be dissolved effectively for the strong deoxidization when the negative potential is lower than -400 mV(vs SHE). Meanwhile, the products can be oxidized easily under higher positive potential, but this reactions are also passivated later. The cathodic reduction is stronger and plays an important role for the dissolution of chalcopyrite. The potentialstatic I��t curve also identify the above conclusion.
Key words: chalcopyrite; decomposition; anodic oxidization; cathodic reduction
��ͭ����һ�ֺ��ѱ������ֽ����������ʽΪCuFeS2, ���ķ���ϵ�����������ÿ�������ӱ�4������(Cu��Fe)������Χ�ơ�ÿ������ͭ���ӱ�4������������Χ������CuΪ+1�ۣ�FeΪ+3�ۡ������и��ʵ����ӵķ����Զ���ǿ���ҹ��ۼ��ļ��ܱȽϴ����ѧ���IJ������ܽϴ�[1]����ͭ��ķֽⷴӦͨ������ת�ƻ��Ѩת�Ʒֲ����У��ҷֽ�Ļ����ϸ���[2]��
��ʵ�ʽ��������У���ͭ��ķֽ�Ҳ�Ƿֲ����еģ��ҷ�Ӧʮ�ֻ������ܶ��о�����Ϊ�жۻ�����ֹ�˷�Ӧ�Ľ�һ�����С������������ͭ��Ĺ����У��ۻ��ﱻ��Ϊ�Ǿ��а뵼�����ʵ��м����ʣ���Cu1-xFe1-yS2-z[3]��CuS2[4-5]��Cu0.8S2 [6]��CuSn[7]�ȡ�����ϸ��������ͭ��Ķۻ�ԭ��һֱ�������飬�ۻ���һ�㱻��Ϊ�Ǹ����ڿ�������Ԫ�ص�����[8]���������[9]���ʡ�������������Щ���ʶ���ۻ����ã��Ƚϻ�ͭ����������ϸ������������ֻ�л�ͭ��ȼ�������Ľ����ʺܵͣ���˵����ͭ������Ķۻ������������еķֽ�����Ƿֲ�����, ����о���ͭ��绯ѧ�ֽ���Ϊ����̽������ϸ�������Ķۻ�ԭ��������塣
���绯ѧ�������о���ͭ��ķֽ�绯ѧ��Ϊ��̽�ֽ����ۻ��������а������ڽ�����ѡ�ȸ��ֵĿ���ӹ�����ǰ�˶Ի�ͭ��ĵ绯ѧ��Ϊ����һЩ�о� [10-14]�����ڴ���Ľ��������������ܽ���̣���˴�����о����ǹ���������������Ӧ������о�[1-7]�����Ի�ͭ��������ԭ��Ӧ����������о����١����о��л�ͭ��缫���þ�ֹ�缫����ת�缫��ѭ������ͼ���бȽϷ�������������Ҫ���ж���ظ�ɨ�裬�ڲ�ͬ��ʼ����ɨ���Լ���ͬ��λ��Χɨ�裬������ͭ���ڳ������������µĵ绯ѧ��Ϊ��
1 ʵ��
1.1���������Լ�
M273���λ�ǣ������ϵͳ(�����ţǣ��ǹ�˾)���������缫ϵͳ: �����缫Ϊ��ͭ����תԲ�̵缫�;�ֹ�缫�����缫��Ϊ�����缫��Ag/AgCl�缫��Ϊ�αȵ缫���ô�³��ëϸ�ܵ������빤���缫��������ʵ���ú���Ϊ99.9%�����������ǡ�
1.2��ʵ�鷽��������
�缫����Ϊ��5 mm��ֱ��Ϊ10 mm��Բ�����״��ͭ�������ĥ�⣬�ñ�ͪ��ϴ����ձ������������缫���������õĿ������Ծ��ķ���ϩΪ���ϵ������������У����û�����֬�ܷ�̶�������缫��¶�Ĺ��������Ϊ0.785 cm2, ��һ���õ���ͭ�����������ӵ���ת���ϣ�������תԲ�̵缫����ֹ�缫��ʾ��ת�ᴦ�ھ�ֹ״̬��
�绯ѧʵ���ں���(298 K)�� pH=2�������½��У�ɨ���ٶ�Ϊ20 mV/s, �缫��ת�ٶ�Ϊ900 r/min�����������Ϊ0.5 mol/L������Na2SO4��Һ��ʵ�������е�λ�������������λ(SHE)���Ա������������й��ڻ�ͭ��ľ���λ��ʾ��������һ�¡�
2 ���������
ͼ1��2��ʾΪ��λɨ�跶Χ��ͬ��ʼ��λ��ͬ��ѭ���������ߡ�ͼ1�����߳�ʼɨ���λ��400 mV��ʼ(��ͭ��ı�����λΪ��=400 mV)��ͼ2�г�ʼ��λΪ-600 mV����ͼ���Ƚ�����ת�缫�;�ֹ�缫�ĵ绯ѧ��Ϊ��ʵ�ߡ����߲��ֱַ������ת����ֹ�缫��ѭ������ͼ��ͨ���Աȿ��������ֵ缫�ĵ����嶼�����棬�����ϴ��������ַ壺���ھ�ֹ�缫���߶���������ת�缫���ߵķ�C1�䡢A3�䡢A4�䣻�������߶����ڣ�������ת�缫���߱Ⱦ�ֹ�缫���ߴ�ķ�A1��C2��C3�����߲��ķ�A0��

ͼ1 ɨ�跶ΧΪ-600~1 000 mV�ͳ�ʼ��λΪ400 mVʱ��ѭ����������
Fig.1 Cyclic voltammograms obtained with chalcopyrite under condition of potential from -600 mV to 1 000 mV and scan potential of 400 mV
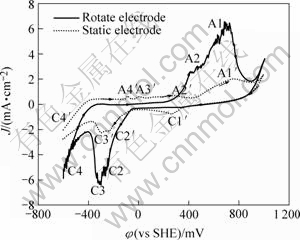
ͼ2 ɨ�跶ΧΪ-600~1 000 mV�ͳ�ʼ��λΪ-600 mVʱ��ѭ����������
Fig.2 Cyclic voltammograms obtained with chalcopyrite under condition of potential range from -600 mV to 1 000 mV and scan potential of -600 mV
1) ����������Ӧ
��һ�������ϸ�������Ĺ����У�����Һ��������ԭ��λԼΪ400~800 mV�����ڴ˵�λ������ͭ���������ֽⷴӦ������ڻ�ͭ��绯ѧ��Ϊ�У��Դ˵�λ���ĵ绯ѧ��Ӧ���о�������Ҫ��ͼ1��ɨ���λ��Ϊ 400~800 mVʱ����ת�缫�;�ֹ�缫�����ϵĵ�����A0��A0�������ƣ��ұ�ʾ�����ܶȶ���С��A0���A0��������Price and Warren[15]�ķ��ִ�����ͬ���ڿ�·��λɨ�赽550 mV�ĵ�λʱ��ѭ������ͼ�ϳ����������壬��Ϊ���������·�Ӧ��
![]()
������������Cu1-xFe1-yS2-z��һ���м������Ӳ��ַֽ���γɵľ��л�ͭ��ṹ��ȱ������,���Կ����������ֽ���ۻ����á�Parker��[16]��Ϊ�������Cu1-xFe1-yS2-z���а뵼�����ԡ�
������A2���е��������ظ�ɨ��������Ӷ����٣�˵�����ǻ�ͭ�����������γɵĵ����塣��A2��A2��ͬ��Ҳ�����÷�Ӧ(1)��ʾ��ֻ������ʱʽ�е�x��y��С���ѡ�
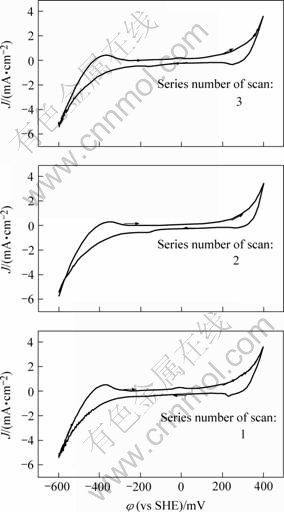
ͼ1��2��ʾ��ֹ�缫�ڵ�λɨ�赽50 mV��-100 mV��������������С������������A3�估A4�䡣��-600~400 mV��λ��Χ�Ķ���ظ�ɨ��ͼ3Ҳ��ʾ��A3���A4������������ظ�ɨ����������Ӷ�������˵�����ǿ��ܲ���ֱ������������Ӧ�����塣��������������Ӧ���ظ�ɨ����������谭��һ��ɨ��Ļ�ѧ��Ӧ����, ���������������⣬ͼ1��2��ʾA3��弰A4�������ֻ�����ھ�ֹ�缫�������У�����ת�缫���߲�û�г��֣��ɴ�˵�����·�Ӧ����������������ԭ���������Ѹ����������[16]��������Ϊ��A3��ͷ�A4���ʾ�ķ�Ӧ��ʽ(2)��ʾ��
![]()

ͼ3 ��ͭ��ֹ�缫��-600~400 mVʱ��ѭ����������
Fig.3 Cyclic voltammograms obtained with static chalcopyrite electrode at potential of -600 mV to 400 mV (1,2,3 are series numbers of scan, scan potential is initiated from -600 mV)
2) ������ԭ��Ӧ
������ѭ������ͼ�У�С��-400 mV��λ�������ڷ�C4�ͷ�C4�䡣Elsherief[17]�����������λɨ�赽-700 mV�����˽ϴ�ԭ���壬��ʱ��ͭ��ֽ������Fe3+�Ѿ�ȫ���ܽ�������������·�Ӧ��
![]()
ʵ�黹�۲����λԽ����C4��ʾ�ĵ�����Խ������Ϊ��C4��C4����ʾ����ʽ(3)��ʾ�Ļ�ѧ��Ӧ��������ԭ��Ӧʹ��ͭ���е�Fe3+�õ�����ΪFe2+�ֽ�����ġ�����������У��µ��м���ﲻ���γɣ�ֱ��Fe3+ȫ���ܽ���������ղ���Ϊͭ�����
ͼ2�е�������ԭ��C2�ͷ�C3ֵԶԶ����ͼ1�е���Ӧ�塣ͼ1�еij�ʼɨ���λΪ400 mV����ͼ2�ij�ʼɨ���λ-600 mV������ͼ2�еķ�C2�ͷ�C3�γ�֮ǰ��ɨ���λ�����˽ϸ���λ�����ɴ�˵�����ϸ���λ�µ�������ԭ�Է�C2�ͷ�C3���γɾ�����Ҫ���á�
Elsherief[17]�ڵ�����м�Fe2+��Cu2+, �����������ӶԻ�ͭ������������Ӱ�졣ͨ���Ի�ͭ��缫���з���ɨ�跢�֣�������ɨ�赽-275 mVʱ�������ԵĻ�ԭ���壬�����Ļ�ѧ��Ӧ�緽��ʽ(4)��(5)��ʾ��
![]()
Elsherief[17]�����������Ӧ����ʽ(4)��(5)��ʾ��ͭ���ֱ�������ֽⷴӦ����ת�缫Ӧ�������ڷ�Ӧ����γɡ���Ȼ����ͼ�г��ֵ�������C2���C3����������ķ�Ӧ��λ��������ǵ缫����תҲ��û����ߵ�����ֵ������ʹ�õ�������ʧ��(��ͼ4)�����Կ����жϷ�C2���C3�䲢����ʾ����(4)��(5)�������ķ�Ӧ��
ͼ4 ��ͭ����תԲ�̵缫��-600~400 mVʱ��ѭ����������
Fig.4 Cyclic voltammograms obtained with rotate chalcopyrite electrode at potential of -600 mV to 400 mV (Arrows indicate potential scan direction. Scan potential is initiated from -600 mV)
ͼ3��ʾΪ������λΪ-300~0 mVʱ���������Ե�������ԭ��C2���C3�䣬����ͼ4�д˵�λ��û�г��ֲ��塣���⣬ͼ3�е�һ��ɨ���ѭ������ͼ�ķ�C2���C3������ص���һ�����壬������ֵ�ϴ�����ɨ��������Ӷ����٣��ɴ�˵���˷�Ӧ�IJ���û�м�ʱǨ�Ƶ���Һ�У��ۻ��ڵ缫�������һ�η�Ӧ��ۻ����á��ٱȽ�ͼ2��4��ͼ2������λ����Ϊ1 000 mV��ͼ4��Ϊ400 mV��ͼ2�еķ�C2��C3�����ԣ���ͼ4����һ��λ��û�й۲��C2��C3����˵������������Ӧ�Ը÷�����Ҳ�ܴ��ɴ˿����Ʋ��C2��C3Ϊ��������������������IJ������������ԭ��Ӧ�����ĵ����塣
�����Ͼ�ֹ�缫��ѭ������ͼ�У�����������ɨ�赽300 mVʱ��������������C1�䡣��ת�缫����û�г�����Щ��, ˵����ʱ�缫��ת�Է�Ӧ�������˾��������á�ͼ5��ʾΪ��ʼɨ�跽��Ϊ��������ɨ�跶ΧΪ0~1 000 mVʱ�ľ�ֹ�缫�ظ�ѭ���������ߡ�ͨ���Ƚ�ͼ1��2����5�ɿ�������C1���������ص㣺���ȣ��˷������еľ�ֹ�缫�϶������ԣ���ʾ��Ӧ�����Ƚϴ���Σ��˷�ֻ�����ھ�ֹ�缫�������ϣ���ת�缫������û�г��֣���˵����ʹ�÷巢���ķ�Ӧ������״ӵ缫�������䣻�ٴΣ������ʼɨ�跽��û�й�ϵ�����ڳ�ʼ��������ɨ��Ҳ�����γ�(��ͼ5)��˵����������������������Ӧ�IJ���Ϊ��Ӧ��ĵ����塣ֵ��ע����ǣ���ͭ������������Ӧ��Ԫ�����ʵ����ɣ��������������Ҳ����������Ӧ�������ۺ����ϼ��㣬������ΪC1����ǵ���Ԫ������������ԭ��Ӧ�Ľ�����ο�����ѧ���������䷴Ӧ�緽��ʽ(6)��ʾ��
![]()

ͼ5 ��ͭ��ֹ�缫��0~1 000 mVʱ�ij�ʼ���������ѭ����������
Fig.5 Cyclic voltammograms obtained with static chalcopyrite electrode at potential of 0~1 000 mV (Arrows indicate scan potential direction, scan potential is initiated from 1 000 mV)
3) ������ԭ�������������
��ͼ1�ɿ���������A0��A0����ͬһ��λ����ѭ����ɨ�����γɺܴ�ķ�A1�ͷ�A1�䣬��ת�缫�����е�A1��Զ���ھ�ֹ�缫��A1��壬�ﵽ4 mA/cm2����˵��һ����缫��תʹ�绯ѧ��ӦŨ��������ü��٣���һ���澭���ϸ���λ��ɨ�跴Ӧ�����ǵ��γ�����Ҫ�����á�
��ͬ��ʼɨ���λ��ѭ������ͼ˵�����������⡣��ͼ6��֪����ʼɨ���λ��300 mV���ϵ���������ʾ�������������ر�С���ҳ�ʼɨ���λԽ����, ����400~800 mV��Χ�IJ���A1��Խ���ɴ˿����Ʋ⣬A1���ʾͣ���ڵ缫�����������ԭ���ﱻ�����������γɵĵ����塣��ͭ���е�Fe3+����ȫ��ԭ�����Ժ���ΪCu2S, ��Ϸ�Ӧ����ʽ(6)����A1��A1������ķ�Ӧ�ɱ�ʾΪ
![]()

ͼ6 ��ͭ����תԲ�̵缫��ͬ��ʼɨ���λ��ѭ����������
Fig.6 Cyclic voltammograms obtained with rotate electrode over different application potential range (Arrows indicate potential scan direction)
Ϊ�˽�һ��֤ʵ��A1��A1����������ԭ��Ӧ�Ĺ�ϵ�Լ����ǶԻ�ͭ��ֽ����Ҫ���壬��������ͬ��ʵ�������£��Ƚ������ʱ���缫�ͺ��λ��=-600 mV��ԭ��ĵ缫ͬ�ں��λ��=550 mV��I��tͼ������ʱ��Ϊ100 s����ͭ���ڦ�=550 mV���λ�·�������������������115 ��A����20 ��A����(��ͼ7(a))����=-600 mV��ԭ�Ժ�ĵ缫�ں��λ��=550 mV����������������4.7 mA����0.2 mA���ң���ֱ���������������˼�ʮ��(��ͼ7(b))������˵����ͭ���ڦ�=550 mV����������������������ԭ���γɵı����м����������ױ�������������Щ���������ѭ���������õ��Ľ�����һ�µġ���ͭ��缫�ڵ�λ��=-600 mV�·�����������ԭ��ӦҲ�Ƚ�ǿ�ң�������-6.3 MA����-2.8 mA����(��ͼ8)��

ͼ7����ͬ��ͭ��缫����Ӻ��λΪ500 mVʱ��I��t����
Fig.7 I��t curves representing deoxidized chalcopyrite at fixed potential of 550 mV: (a) Fresh electrode; (b) Reducted electrodes over potential -600 mV
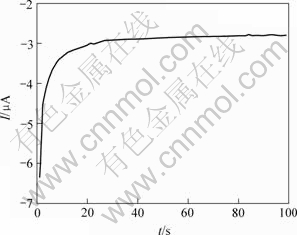
ͼ8 ��ͭ������Ӻ��λΪ-600 mV��I��t����
Fig.8 I��t curve representing reduction of chalcopyrite at fixed potential of -600 mV
3 ����
1) �ڳ������������£���ͭ�����������������������Ϊ�ֽ�������м�ۻ�����͵���Ԫ�������ɡ�
2) ��һ����Ӹ���λ�£���ͭ��������ԭ��Ӧ������������ӦҪǿ�ҵö࣬�������ڵ�λС��-400 mV������¡�����������λ�ﵽһ��ֵ�� Fe3+����ȫ��ԭ�������γ��ȶ��Ĺ������Cu2S�������м����ʲ��������䣬����������λ���ܷ����Ͽ������������Ӧ������������γɶۻ���CuxS��
REFERENCES
[1] ��־��. ͭ���տ���ѧ[J]. ������ɫ����ѧ��, 1999, 9(1): 1-8.
XU Zhi-hua. Technological mineralogy of copper[J]. Journal of Guangdong Non-Ferrous Metals, 1999, 9(1): 1-8.
[2] �����, ������. ��ϸ�������İ뵼���ܴ����۷���[J]. ��ɫ����, 2004, 56(3): 36-48.
LI Hong-xu, WANG Dian-zuo. Fundamental analysis of sulfide bioleaching process based on semiconductor electrochemistry[J]. Nonferrous Metals, 2004, 56(3): 36-48.
[3] Warren G W, Wadsworth M E, EI-Raghy S M. Anodic behavior of chalcopyrite in sulfuric acid[J]. Metallurgical Soc of AIME, 1982, 13(4): 261-275.
[4] Vaughan D J. Atmospheric and electrochemical oxidation of the surface of chalcopyrite[J]. Geochimica et Cosmochimica Acta, 1995, 59(1): 1091-1100.
[5] Yin Q, Vaughan D J, Kelsall G H, England K E R. Electrochemical oxidation of covellite(CuS) in alkaline solution[J]. Journal of Colloid and Interface Science, 1994, 59(6): 133-142.
[6] Klauber C, Parker A, Bronswijk W, Watling H R. Sulphur speciation of leached chalcopyrite surfaces as determined by X-ray photoelectron spectroscopy[J]. Mineral Process, 2001, 62(1): 65-94.
[7] Hack R P, Dreisinger D B, Peters E, King J A. Passivation of chalcopyrite during oxidative leaching in sulfate media[J]. Hydrometallurgy, 1995, 39(3): 25-49.
[8] Dutrizac J E. Elemental sulphur formation during the ferric chloride leaching of chalcopyrite[J]. Hydrometallurgy, 1990, 23(2): 153-176.
[9] Stott M B, Watling H R, franzmann P D, Sutton D. The role of iron-hydroxy precipitates in the passivation of chalcopyrite during bioleaching[J]. Mineral Engineering, 2000, 13(1): 1117-1127.
[10] Woods R, Hope G A. Spectroelectrochemical investigation of the intersection of o-isopropyl-nethylthionocarbomate with copper surfaces[J]. Colloids Surf, 1999, 146(1): 63-74.
[11] Buckley A N, Woods R. Under potential deposition of dithiophosphate on chalcocite[J]. Electroanal Chem, 1993, 357(1): 387-405.
[12] Koch D F A, McIntyre R. The application of reflectance spectroscopy to a study of the anodic oxidation of cuprous sulphide[J]. Electroanal Chem, 1976, 71(1): 285-296.
[13] �����, �����. ԭ���ЧӦ�Ի����ϸ��������Ӱ��[J]. �й���ɫ����ѧ��, 2000, 13(5): 1284-1287.
LI Hong-xu, QIU Guan-zhou. Galvanic effect on mixed sulfide bioleaching[J]. The Chinese Journal of Nonferrous Metals, 2000, 13(5): 1284-1287.
[14] ŷ����, ������. ��ѡ�����л�ͭ�����Ƶĵ绯ѧ�о�[J]. ��ұ����, 1999, 19(3): 68-86.
OU Le-ming, FENG Qi-ming. A study on electrochemistry of depressing chalcopyrite in flotation[J]. Mining and Metallurgical Engineering, 1999, 19(3): 68-86.
[15] Price D W, Warren G W. The influence of silver ion on the electrochemical response of chalcopyrite and other mineral sulfide electrodes in sulfuric acid[J]. Hydrometallurgy, 1986, 15(1): 303-324.
[16] Parker A J, Paul R L, Power G P. Electrochemistry of the oxidative leaching of copper from chalcopyrite[J]. Electroanal Chem, 1981, 118(1): 305-316.
[17] Elsherief A E. The influence of cathodic reduction Fe2+ and Cu2+ ions on the electrochemical dissolution of chalcopyrite in acidic solution[J]. Mineral Engineering, 2002, 15(1): 215-223.
������Ŀ�������ص�����о���չ�滮������Ŀ(2004 CB619204)
�ո����ڣ�2006-04-10�������ڣ�2006-06-26
ͨѶ���ߣ�¬�����������ڣ���ʿ�о������绰��0731-8830913��E-mail: Luyp309@sohu.com
