
���ܴ�����AP65þ�Ͻ������ŵ���Ե�Ӱ��
���˹⣬���ճ�������Ⱥ�����ޣ�ʯ�������ϲ
(���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ�����ϣ���ɳ 410083)
ժ Ҫ��
�Ʊ�AP65þ�Ͻ��������ϣ�����ɨ��羵(SEM)���������(ESA)�о���ͬ״̬������������֯��ʴ��ò������X��������(XRD)������ͬ״̬����������ṹ�����ö���λ����ɨ�跨��������������ͽ����迹���о���ͬ״̬��������3.5% NaCl��Һ�еĵ绯ѧ��Ϊ���о������������̬�����ھ��紦���ڲ������ֲ��ĵڶ���Mg17Al12���õڶ�������ǿ��������ʴ�ԣ������������ֲ���ʴ�����ҷŵ���Խ�������400 �����24 h��ڶ����ܽ⣬��������ʴ�Լ�С����ʴ�����̬���������ҷŵ������ǿ��
�ؼ��ʣ�
þ������AP65�Ͻ����绯ѧ��������ʴ��������������������迹��
��ͼ����ţ�TG146.1+5 ���ױ�־�룺A ���±�ţ�1672-7207(2012)06-2120-05
Effect of solid solution treatment on discharge activity of AP65 magnesium alloy anode
WANG Nai-guang, WANG Ri-chu, PENG Chao-qun, FENG Yan, SHI Kai, JIN He-xi
(School of Materials Science and Engineering, Central South University, Changsha 410083, China)
Abstract: AP65 magnesium alloy anode was prepared by melting and casting. The microstructures and corrosion morphologies of the specimens under different conditions were investigated by scanning electronic microscopy (SEM) and emission spectrum analysis (ESA), the phase structures of the specimens under different conditions were analyzed by X-ray diffraction (XRD), and the electrochemical behavior of the specimens under different conditions in 3.5% NaCl solution was studied by potentiodynamic polarization, galvanostatic oxidation test and electrochemical impedance spectroscopy. The results show that the second phase Mg17Al12 distributing discontinuously along the grain boundaries of the as-cast specimen can enhance the corrosion resistance but accelerate the local corrosion or reduce the discharge activity of AP65 anode. After solid solution treatment at 400 �� for 24 h, the second phase dissolves into the magnesium matrix and the corrosion resistance decreases but the corrosion develops uniformly on the specimen surface, leading to an improvement of the activity of AP65 anode.
Key words: magnesium anode; AP65 alloy; electrochemical activity; corrosion resistance; galvanostatic test; electrochemical impedance spectroscopy
þ�Ͻ��������ϵĺ�ˮ�������ؾ��е绯ѧ���Ըߡ���ѹ��Χ�㡢�����ܶȴ�δ������ʱ����ʱ�䳤[1-2]���ص㣬�㷺Ӧ���ں�����������װ�á�DZˮͧ�����渡�ꡢ��״��ء��ռ�������ͽ��������豸������[3]����ˮ���������������(��þ�Ͻ�����)�ں�ˮ�е��ܽ��ṩ�����ŵ�������õ���ͨ����ӵ�·����������Ȼ���������Ϸ�����ԭ��Ӧ����ˮ�����ͻ�����ص��Dz���ҪЯ������ʣ������ڷŵ�ʱ������Ȼ��ˮ��Ϊ���Һ������������о���ȫ[4]����þ�Ͻ��������ϴ��ڼӹ��ѡ��Ը�ʴ���ʴ���ʱ�䳤�ҵ���Ч�ʵ͵�ȱ��[5-6]��Ŀǰ�������������ķ����Ǹ����ȴ����ƶȺ����������ĺϽ�Ԫ�ء�AP65��þ�Ͻ������е�һ�֣�������ɷ�ΪMg-6%Al-5%Pb(��������)��AlӰ��þ�����ķŵ���ԣ���Ч��ȡ����Al�ĺ����͵ڶ���ķֲ�[7-8]��������Pb������ǿþ�����ķŵ����[9]��Udhayan��[10]�о�AP65�����ڸ�����þ��Һ�еĵ绯ѧ��Ϊ��������缫��Ӧ�����ܻ���ơ��о��������[11]��Al��Pb��Mg����Ļ����ЭͬЧӦ����NaCl��Һ�зŵ�ʱ�ܽ��Pb2+�����������ʽ��Mg�缫����������ù����������ܽ��Al3+������Al3+��Al(OH)3����ʽ������Mg�缫���棬��Al(OH)3��2Mg(OH)2����ʽ���븯ʴ����Ĥ���Ե缫����á�Ŀǰ����������ڲ�ͬ״̬��AP65������NaCl��Һ�е绯ѧ��Ϊ�ı������٣�Ϊ�ˣ����������о���̬����̬AP65������3.5% NaCl��Һ�еĵ绯ѧ��Ϊ���ԱȽϲ�ͬ״̬��AP65�����ķŵ����ܲ��������ܴ�����AP65�����ŵ���Ե�Ӱ�졣
1 ʵ��
���ø�Ӧ�������Ʊ�AP65þ�Ͻ��������ϣ���Mg��Al��Pb���Դ���Ϊ99.99%�Ľ�������ߴ�ʯī��������750 ������������������������������������������ģ����������������400 �����24 h��ˮ�㡣����ԭ�����չ�����AP65�����Ļ�ѧ�ɷ�(��������)������1�� ����Quanta-200ɨ��羵����������о�����ĥ����������������֯������D/Max2500�����Ƿ�����̬����̬AP65��������ṹ����ɨ���ٶ�Ϊ1.2 (��)/min��ɨ�跶ΧΪ10��~80�㣬����Cu�У�������ѹΪ47 V����������Ϊ250 mA��
��1 AP65�����Ļ�ѧ�ɷ�(��������)
Table 1 Chemical composition of AP65 anode %

���ö���λ����ɨ�跨��������������ͽ����迹���ⶨAP65�����ĵ绯ѧ��Ϊ����������ͬ�ͺ�SiCɰֽ��ĥȥ�����������㣬�����侭��ĥ�Ĺ����沢ʹ���10 mm��10 mm(������)�ľ��Ρ�Ȼ����ͭ����������Ʒ���ǹ������û�����֬�ܷ⡣�绯ѧ����ΪIM6ex���������缫��ϵ���в����������缫ΪAP65�����������缫Ϊ���缫���αȵ缫Ϊ���ʹ��缫��ʵ���¶�Ϊ25 �棬���ҺΪ3.5%(��������)��NaCl������Һ���õ��Һ�ӽ���ˮ�ɷ֡�����λ����ɨ����Ե�ɨ���ٶ�Ϊ2 mV/s����ѹ��ΧΪ��·��λ��0.8 V��������������Եĵ����ܶ�Ϊ180 mA/cm2���ŵ�ʱ��Ϊ600 s�������迹���ԵĹ�����λΪ��·��λ��Ƶ�ʷ�ΧΪ100 kHz��0.05 Hz����ѹ���Ϊ5 mV�����绯ѧ����������������Һ������ ���С�
����ԭ�����չ��ײⶨ��������������������Ժ��ܽ��Mg2+�ͺϽ�Ԫ�����ӵ�Ũ�ȡ�����Quanta-200ɨ��羵�۲��������������������Ժ�ĸ�ʴ������ò(����ʴ����Ͳ�����ʴ����)��������3.5% NaCl��Һ�н���48 h��ĸ�ʴ���档����200 g/L CrO3+10 g/L AgNO3��Һ����������ĸ�ʴ������������ʴ�������������ˮ�Ҵ��г�������ϴ5 min�����ﲢ�۲�������ĸ�ʴ��ò��
2 ��������
2.1 ���ܴ�����AP65��������֯����ṹ��Ӱ��
ͼ1��ʾΪ��ͬ״̬��AP65������XRDͼ�ס���ͼ1(a)��֪����̬�����ɦ�-Mg����͵ڶ���Mg17Al12��ɣ�����̬��������Ҫ����-Mg����(ͼ1(b))��ͼ2��ʾΪAP65������̬�����ı�ɢ���Ͻ�Ԫ�ص���ֲ�����ͼ2(a)��ʾ�ı�ɢ������Կ�������̬�����еڶ���Mg17Al12��Ҫ�ھ��紦�������ֲ������ڵڶ�����١���ͼ2(b) ��ʾMg����ֲ���֪��Mg��Ҫ�ֲ��ھ��ڣ����紦Mg�������١���ϱ�2����������λ�ھ����A��Mg����Ϊ61% (������������ͬ)������B��Ϊ93.71%����ͼ2(c) ��ʾAl����ֲ����Կ�����Al��Ҫ�ھ��紦ƫ�ۣ����� Al�������١���ϱ�2��֪��λ�ھ���A��Al����Ϊ35.29%����������C��Ϊ10.25%������B��Ϊ2.70%��ͼ2(d)��ʾΪPb����ֲ������п��Կ���Pb�����Al�ֲ��Ͼ��ȡ���ϱ�2��֪����Pb�ĺ���ƫ���С��
ͼ3��ʾΪAP65��������̬�����ı�ɢ���Ͻ�Ԫ�ص���ֲ�����ͼ3(a)��ʾ�ı�ɢ������Կ���������̬������ҪΪ����-Mg����ĵ�����֯��������̬������400 �����24 h��ڶ�����������塣����ͼ3(b)~(d) ��ʾ���Ͻ�Ԫ�ص���ֲ���֪�������ܴ�������Ͻ�Ԫ�������̬�����ֲ��Ͼ��ȡ���ϱ�2����������ͼ3(a)��D�����Ͻ�Ԫ�صĺ����ӽ���AP65������ɷ֡�
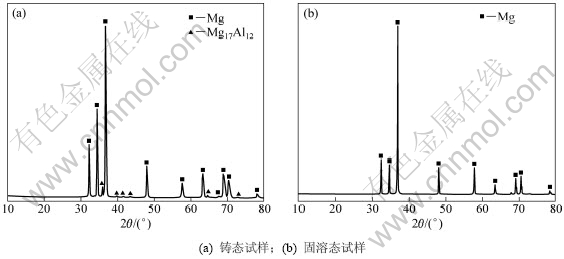
ͼ1 ��ͬ״̬��AP65������XRDͼ��
Fig.1 XRD patterns of AP65 anodes under different conditions

ͼ2 AP65������̬�����ı�ɢ���Ͻ�Ԫ�ص���ֲ�ͼ
Fig.2 BSE (Backscattered electron) images of as-cast specimen of AP65 anode and elemental mappings of alloying elements
2.2 ���ܴ�����AP65�����������ߺͺ�������������еĵ�λ-ʱ�����ߵ�Ӱ��
ͼ4(a)��ʾΪ��ͬ״̬��AP65�����ļ������ߡ�������������֧������֧���Գƣ�����֧�����ܶ����λ���ӵ����ʸ�������֧��һ����˵������֧�ĵ�����Ҫ��������ƣ�����֧�ĵ�����Ҫ�ܽ����������ܽ����[12]���ڶ���λ����ɨ������У���������û�з����ۻ������ݼ�����������������ĸ�ʴ��λ��ʴ�����ܶȣ�����3���ӱ�3���Կ���������̬�����ĸ�ʴ��λΪ-1.428 V��������̬����(-1.598 V)������̬�����ĸ�ʴ�����ܶ�Ϊ0.531 mA/cm2��������̬�����ĵ����ܶ�(0.225 mA/cm2)��þ�Ͻ��������ձ���ڵ�ż��ʴ���ڶ����þ������и��ߵĵ缫��λ���䵱����[5]����ֲ��������Ե绯ѧ��ʴ��Ϊ�кܴ�Ӱ�죻���ڶ������������ʱ����Ҫ��Ϊ��������þ�����γɸ�ʴ��ż����þ����ĸ�ʴ�����ڶ���������϶�ʱ������Ҫ��Ϊ��ʴ��������þ����ĸ�ʴ[13]�����ͼ1��ʾ��XRDͼ��ͼ2��ʾ������֯��֪����̬�������紦���ڲ������ֲ��ĵڶ���Mg17Al12���õڶ������þ����Ϊ�������࣬����þ���帯ʴ��Ч���ϲ�[8]��AP65������̬�����ĸ�ʴ�����ܶȱȹ���̬��С��������̬�����еڶ���Mg17Al12�����ƻ���ĸ�ʴ������ͼ4(a)����ͬһ������λ��(������ʾ)����̬���������������ܶȱȹ���̬��С�������ڶ���Mg17Al12������������Ӧ������������������

ͼ3 AP65��������̬�����ı�ɢ���Ͻ�Ԫ�ص���ֲ�ͼ
Fig.3 BSE (Backscattered electron) images of solid solution treated specimen of AP65 anode and elemental mappings of alloying elements
��2 ͼ1��ͼ2�о����������ø�����Ļ�ѧ�ɷ� (��������)
Table 2 Chemical compositions of different areas in Figs.1-2 obtained by ESA�� ������ %
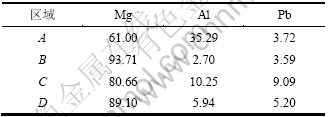
ͼ4(b) ��ʾΪ��ͬ״̬��AP65�����ں�������������еĵ�λ-ʱ�����ߡ��ú���������������������������������²�ã� �����ܶ�Ϊ180 mA/cm2���ں�������������У���ɫ��ʴ���ﲻ�ϴ�����������䡣���ݵ�λ-ʱ�����߿ɼ����������ƽ����λ������3��һ����˵��ƽ����λ�ܷ�ӳþ�����ķŵ���ԣ�ƽ����λԽ������ŵ����Խǿ����ͼ4(b)���Կ�������̬�����ĵ�λ-ʱ�����ߴ��ڲ��������ƽ����λ����(-1.516 V)������̬������ŵ�ƽ����ƽ����λ�ϸ�(-1.683 V)����ˣ�����̬���������̬���н�ǿ�ķŵ���ԡ�ͼ4(c) ��ʾΪ��ͬ״̬��AP65�����������������λ��������������ܽ��Mg2+Ũ�ȣ�����̬������Mg2+Ũ��Ϊ72.4 mg/(cm2��L-1)��������̬������Mg2+Ũ��(51.3 mg/(cm2��L-1)�����������ܴ�����AP65�ڷŵ������þ����������ܽ��ٶȼӿ졣ͼ4(d)��ʾΪ��ͬ״̬��AP65�����������������λ��������������ܽ��Al3+��Pb2+Ũ�ȣ�����̬�����ܽ��Al3+Ũ�ȴ�����̬������Ũ�ȣ���Pb2+Ũ����С����̬����Ũ�ȡ���������[11]��AP65������NaCl��Һ�зŵ�ʱ�ܽ��Pb2+�����������ʽ��Mg�������������ù����������ܽ��Al3+�ij�����Al3+��Al(OH)3����ʽ������Mg������棬��Al(OH)3��2Mg(OH)2����ʽ���븯ʴ����Ĥ���Ի�������á�һ����˵��þ�����ĵ绯ѧ����ȡ���������ijɷֺͽṹ����̬�����кϽ�Ԫ�طֲ��������ҵڶ���Mg17Al12�����ƻ���ĸ�ʴ����ˣ���绯ѧ���Խ����������ܴ�����ڶ����ܽ��ҺϽ�Ԫ����ɢ����(��ͼ3)���ڷŵ�������ܽ��Al3+Ũ�����������ڸ�ʴ����Ĥ�İ��䣬��ˣ���绯ѧ���Խ�ǿ���ں��������������ƽ����λ�ϸ���Mg������ܽ��ٶȼӿ졣
��3 ��ͬ״̬��AP65�����ĸ�ʴ��λ(Ecorr)����ʴ�����ܶ�(icorr)��ƽ����λ(Emean)
Table 3 Corrosion potentials (Ecorr), corrosion current densities(icorr) and mean potentials (Emean) of AP65 anodes under different conditions

ͼ4 ��ͬ״̬��AP65�����Ķ���λ����ɨ�����ߡ���������ߡ���������Ժ�������ܽ��Mg2+����Ũ�ȺͺϽ�Ԫ������Ũ��
Fig.4 Potentiodynamic polarization curves��potential-time curves, concentration of dissolved Mg2+ ions and
alloying elements ions of AP65 anodes under different conditions after galvanostatic tests
2.3 ���ܴ�����AP65�����绯ѧ�迹��Ӱ��
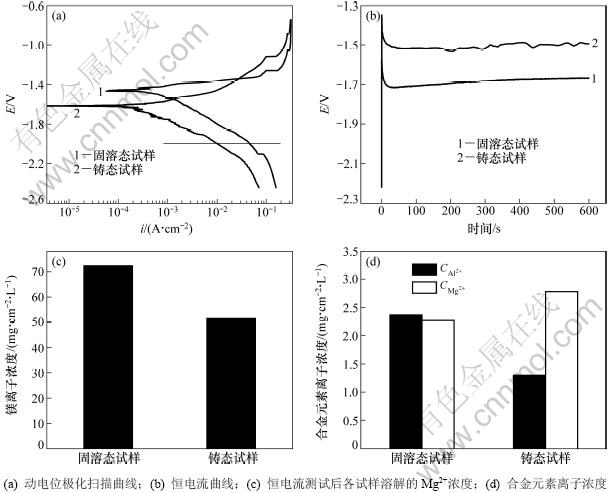
ͼ5��ʾΪ��ͬ״̬��AP65�����ĵ绯ѧ�迹��(���У�Z��Ϊ�迹ʵ����Z��Ϊ�迹�鲿)�����迹���������Ŀ�·��λ�²�ã��ܷ�ӳ���������Ը�ʴ״̬�µĵ绯ѧ��Ӧ����������ͼ5(a)��ʾ��Nyquistͼ��֪����̬����������Ƶ�ʷ�Χ�ڽ�����1���ݿ��������ݿ�����˫������Cdl�͵��ת�Ƶ���Rt�йأ�����Ӱ����̬�����缫���淴Ӧ��״̬����Ϊ�缫��λE[14]������̬���������ڸ�Ƶ������1����˫������Cdl�͵��ת�Ƶ���Rt�йص��ݿ����⣬�ڵ�Ƶ��Ҳ����1���뾶��С���ݿ��������ݿ������������������ĸ�ʴ����Ĥ�йأ�����ݺ͵���ֱ���Cf��Rf��ʾ����ˣ�Ӱ�����̬�����缫���淴Ӧ��״̬����Ϊ�缫��λ�ͳ����ڱ���ĸ�ʴ����Ĥ[15]���ø�ʴ����Ĥ��Ҫ�ɷ�ΪMg(OH)2�����ܽ��Mg2+Ũ�ȳ���ijһ����ֵʱ������Mg(OH)2����ʽ�������������[14]�����ݵ绯ѧ�迹��ԭ��[15]�����缫���淴Ӧ���ܵ缫��λӰ��ʱ�������ڵ���YF�ɱ�ʾΪ��
YF=1/Rt (1)
ʽ�У�YFΪ�����ڵ��ɣ�RtΪ���ת�Ƶ��衣���缫���淴Ӧ�����ܵ缫��λӰ���⣬���ܳ����ڱ���ĸ�ʴ����ĤӰ��ʱ�������ڵ���YF�ɱ�ʾΪ��
YF=1/Rt+(?IF/?��)( ?�ȡ�/?E)/[j��-?�ȡ�/?��] (2)
ʽ�У�IFΪ�����ڵ����ܶȣ���Ϊ��ʴ����Ĥ����������ĸ����ʣ�EΪ�缫��λ����ΪƵ�ʣ�![]() ���ȡ�=d��/dt��tΪʱ�䡣
���ȡ�=d��/dt��tΪʱ�䡣
ͼ5 ��ͬ״̬��AP65�����ĵ绯ѧ�迹��
Fig.5 EIS of AP65 anodes under different conditions
�ڵ缫��Ӧ�����У������ڵ����ܶ�IF�港ʴ����Ĥ�����ʵ��������С����ˣ�?IF/?�ȣ�0����缫��λ�������ܽ��Һ�е�Mg2+Ũ��������Mg(OH)2Ĥ�����������������ˣ�?�ȡ�/?E��0������ʽ(2)��(?IF/?��)( ?�ȡ�/?E)��0�������ڵ�Ƶ��������1���ݿ���[14]����ʵ�������������ͼ5(b)��ʾ��Bodeͼ��֪����̬�������迹ģ������Ƶ�ʷ�Χ�ڶ����ڹ���̬���迹ģ��������̬���������̬��Ⱦ��н�ǿ����ʴ�ԡ����ݵ绯ѧ�迹����ϳ��������ĵ绯ѧԪ����������4���������������ĵ�Ч��·ͼ����ͼ6���ڵ�Ч��·ͼ�У�RsΪ����������³��ëϸ��֮�����Һ���衣������ɢЧӦ�Ĵ��ڣ����ó���λ��Ԫ��CPEdl����˫������Cdl����Ԫ������ת�Ƶ���Rt�������ӱ�4���Կ���������̬�����ĵ��ת�Ƶ���Ϊ325 ����cm2������̬����(760 ����cm2)��С����������̬��������̬��Ⱦ��н�ǿ�ĵ绯ѧ���ԡ���ˣ�����̬������NaCl��Һ�н���ʱ�����̬�����и����Mg2+�ܽ��Һ�У���Mg2+��Ũ�ȳ���ijһ����ֵʱ������Mg(OH)2����ʽ����������� ��[15]�����¹���̬������Nyquistͼ�ڵ�Ƶ�������ݿ���[14]��
2.4 ���ܴ�����AP65������ʴ��ò��Ӱ��
ͼ7��ʾΪ��ͬ״̬��AP65������������������Ժ�ʴ������ò�Ķ��ε�����ͼ7(a)��(b)���Կ��������������������������涼����һ�㸯ʴ����ø�ʴ����������Ҵ�������״���ƣ��ڴ�����ܶȷŵ���������״�����������䣬�Ի������������á�ͼ7(c)��ʾΪ��̬����������������������ʴ����ı�����ò�����Կ�����̬������ʴ���氼��ƽ�Ҳ��ֵڶ����ڷŵ���������䡣����ͼ7(d)��ʾ�Ŵ�Ķ��ε������֪����̬�����ں�������������л���ĸ�ʴ�ӵڶ�����Χ��ʼ��ͼ7(e)��ʾΪ����̬��������������Ժ������ʴ����ı�����ò�����п��Կ�������̬������ʴ���������̬������ƽ̹�����ڻ������ľ��ȸ�ʴ��ͼ8��ʾΪ����������48 h��ʴ����ĵͱ���ɢ����ͼ8(a)���Կ�������̬������ʴ������ڽϴ�İ��ӣ��ֲ���ʴ���ء�����̬������ʴ����(��8(b))����̬�������ܸ�ʴ����С�ڹ���̬�������ڵڶ���Ĵ��ڣ����¾ֲ���ʴ�����һ��Խϲ���ʺ����ڴ��ʺ�ˮ����������ϡ�������̬�������ڳɷ־��ȣ��ں�����������������������ܽ⣬�һ��Խ�ǿ���ʺ����ڴ��ʺ�ˮ����������ϡ�
��4 ���ݵ绯ѧ�迹����ϳ��IJ�ͬ״̬��AP65�����ĵ绯ѧ����
Table 4 Electrochemical parameters obtained by fitting analysis of EIS of AP65 anodes under different conditions

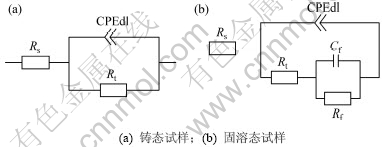
ͼ6 ��ͬ״̬��AP65�����ĵ�Ч��·ͼ
Fig.6 Equivalent circuits for EIS of AP65 anodes under different conditions
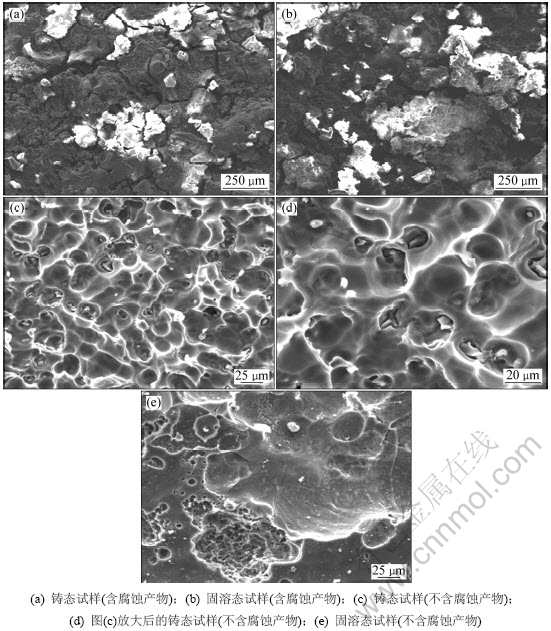
ͼ7 ��ͬ״̬��AP65��������������Ժ�ʴ������ò�Ķ��ε�����
Fig.7 SE (Secondary electron) corrosion morphologies of AP65 anodes under different conditions after galvanostatic tests

ͼ8 ��ͬ״̬��AP65������3.5% NaCl ��Һ�н���48 h��ʴ����ı�ɢ����
Fig.8 BSE (Backscattered electron) cross-section images of AP65 anodes immersed in 3.5% NaCl solution for 48 h
3 ����
(1) AP65������̬�����Ͻ�Ԫ�طֲ����������ھ��紦���ڲ������ֲ��ĵڶ���Mg17Al12���õڶ�������ǿ��������ʴ�ԣ������������ֲ���ʴ�����ҷŵ���Խ��������ʺ��������ʺ�ˮ����������ϡ�
(2) AP65��������̬����Ϊ������֯�ҺϽ�Ԫ�طֲ��Ͼ��ȣ�����ʴ�Ա���̬������С����ʴ�����̬���������ҷŵ���Խ�ǿ����180 mA/cm2�ŵ�����ܶ���ƽ����λΪ-1.683 V�������ڴ��ʺ�ˮ����������ϡ�
�ο����ף�
[1] Medeiros M G, Dow E G. Magnesium-solution phase catholyte seawater electrochemical system[J]. Journal of Power Sources, 1999, 80(1/2): 78-82.
[2] Renuka R. Influence of allotropic modifications of sulphur on the cell voltage in Mg-CuI(S) seawater activated battery[J]. Materials Chemistry and Physics, 1999, 59(1): 42-48.
[3] Renuka R. AgCl and Ag2S as additives to CuI in Mg-CuI seawater activated batteries[J]. Journal of Applied Electrochemistry, 1997, 27(12): 1394-1397.
[4] Kim J G, Joo J H, Koo S J. Development of high-driving potential and high-efficiency Mg-based sacrificial anodes for cathodic protection[J]. Journal of Materials Science Letters, 2000, 19(6): 477-479.
[5] ���˹�, ���ճ�, ����, ��. �Ͻ��ȴ�����þ�Ͻ�����������֯�����ܵ�Ӱ��[J]. �й���ɫ����ѧ��, 2009, 19(1): 38-43.
WANG Nai-guang, WANG Ri-chu, YU Kun, et al. Effect of alloying and heat treatment on electrochemical behavior of Mg anode[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(1): 38-43.
[6] Flamini D O, Saidman S B, Bessone J B. Aluminium activation produced by gallium[J]. Corrosion Science, 2006, 48(6): 1413-1425.
[7] GU Xue-nan, ZHENG Yu-feng, CHENG Yan, et al. In vitro corrosion and biocompatibility of binary magnesium alloys[J]. Biomaterials, 2009, 30(4): 484-498.
[8] ZHAO Ming-chun, LIU Ming, SONG Guang-ling, et al. Influence of the ��-phase morphology on the corrosion of the Mg alloy AZ91[J]. Corrosion Science, 2008, 50(7): 1939-1953.
[9] Candan S, Unal M, Turkmen M, et al. Improvement of mechanical and corrosion properties of magnesium alloy by lead addition[J]. Material Science and Engineering A, 2009, 501(1/2): 115-118.
[10] Udhayan R, Bhatt D P. On the corrosion behavior of magnesium and its alloys using electrochemical techniques[J]. Journal of Power Sources, 1996, 63(1): 103-107.
[11] WANG Nai-guang, WANG Ri-chu, PENG Chao-qun, et al. Influence of aluminium and lead on activation of magnesium as anode[J]. Trans Nonferrous Met Soc China, 2010, 20(8), 1403-1411.
[12] Tamar Y, Mandler D. Corrosion inhibition of magnesium by combined zirconia silica sol-gel films[J]. Electrochimica Acta, 2008, 53(16): 5118-5127.
[13] SONG Guang-ling, Andrej A, Matthew D. Influence of microstructure on the corrosion of diecast AZ91D[J]. Corrosion Science, 1998, 41(2): 249-273.
[14] �ܳ���. ��ʴ�绯ѧԭ��[M]. ����: ��ѧ��ҵ������, 2008: 150-160.
CAO Chu-nan. Principles of electrochemistry of corrosion[M]. Beijing: Chemical Industry Press, 2008: 179-187.
[15] ZHAO Ming-chun, LIU Ming, SONG Guang-ling, et al. Influence of pH and chloride ion concentration on the corrosion of Mg alloy ZE41[J]. Corrosion Science, 2008, 50: 3168-3178.
(�༭ �²ӻ�)
�ո����ڣ�2011-07-15�������ڣ�2011-08-22
������Ŀ����������ҹ�����Ŀ(JPPT-115-168)
ͨ�����ߣ����ճ�(1965-)���У��㶫��ƽ�ˣ���ʿ�����ڣ����º�ˮ���������о����绰��0731-88836638��E-mail��wrc910103@163.com
ժҪ�������������취�Ʊ�AP65þ�Ͻ��������ϣ�����ɨ��羵(SEM)���������(ESA)�о���ͬ״̬������������֯��ʴ��ò������X��������(XRD)������ͬ״̬����������ṹ�����ö���λ����ɨ�跨��������������ͽ����迹���о���ͬ״̬��������3.5% NaCl��Һ�еĵ绯ѧ��Ϊ���о������������̬�����ھ��紦���ڲ������ֲ��ĵڶ���Mg17Al12���õڶ�������ǿ��������ʴ�ԣ������������ֲ���ʴ�����ҷŵ���Խ�������400 �����24 h��ڶ����ܽ⣬��������ʴ�Լ�С����ʴ�����̬���������ҷŵ������ǿ��
