

Evaluation of reliability of Coats-Redfern method for kinetic analysis of non-isothermal TGA
R. Ebrahimi-Kahrizsangi1, 2, M. H. Abbasi2
1. Department of Engineering, Islamic Azad University, Najafabad branch, Isfahan, Iran;
2. Department of Materials Engineering, Isfahan University of Technology, Isfahan, Iran
Received 18 April 2007; accepted 31 July 2007
Abstract:
A critical examination was made on the reliability of kinetic parameters of nonisothermal thermoanalytical rate measurement by the widely applied Coats-Redfern(CR) equation. For this purpose, simulated TGA curves were made for reactions with different kinetic models, including chemical, diffusion (Janders) and mixed mechanism at different heating rates. The results show that, for reactions controlled kinetically by one mechanism, all solid state reaction models show linear trends by use of CR method and this method can not distinct the correct reaction model. For reactions with mixed mechanism, the CR method shows nonlinear trends and the reaction models and kinetic parameters can not be extracted from CR curves. The overall conclusion from this comparative appraisal of the characteristics of the CR approach to kinetic analysis of TGA data is that the CR approach is generally unsuitable for determination of kinetic parameters.
Key words:
kinetic analysis; Coats-Redfern equation; nonisothermal TGA; rate equation; Arrhenius parameters;
1 Introduction
Kinetic analysis of thermal decomposition processes has been the subject interest for many investigators all along the modern history of thermal decomposition. The interest is fully justified. On one side, kinetic data are essential for designing any kind of device, in which the thermal decomposition takes place; on the other side, kinetics is intrinsically related with the decomposition mechanisms. The knowledge of the mechanism allows the postulation of kinetic equations or vice versa, and kinetics is the starting point to postulate mechanisms for the thermal decomposition[1].
Although kinetic studies can be performed in different devices, thermogravimetry(TG) is, by large, the mostly used technique. This technique consists of preheating the sample to a given temperature (T0) and then starting the experiment with a fixed nominal heating rate (��). So, theoretically it is possible to write
T=T0+��?t (1)
So in a TG experiment, a modern equipment typically registers hundreds or thousands of experimental points that can be used for kinetic analysis of the reaction.It is clear that the selection of correct model is a critical point in kinetic analysis. Knowing how a model can justify experimental data has been evaluated by many researchers[1-3]. There are different methods to study the kinetics of non-isothermal processes. These include statistical methods[4-8], predictions of activated complex theory for the value of the pre-exponential factor[9], methods based on the fact that, for different reaction models, the extent of reaction at maximum reaction rate amax falls into a narrow specific range[10], Coats-Redfern (CR) method[11] and iso-conversional model free methods[12].
2 Rate equations
Usually the change in extent of reaction (��) is used to study the solid state reactions kinetics:
![]() (1)
(1)
where m0, mt and m�� are initial sample mass, sample mass at time t and sample mass at the end of reaction, respectively.
Using extent of reaction, the rate of a solid state reaction can be generally described by
![]() (3)
(3)
Integration of the above equation gives the integral rate law:
g(��)=kt (4)
Several reaction models[13] using f(��) or g(��) are listed in Table 1.
Table 1 Solid state rate equations

The explicit temperature dependence of the rate constant is introduced by replacing k(T) with the Arrhenius equation which gives
![]() (5)
(5)
And
![]() (6)
(6)
where A (the pre-exponential factor) and Ea (activation energy) are the Arrhenius parameters. These parameters together with the reaction model are sometimes called the kinetics triplet. Under non-isothermal conditions, in which a sample is heated at a constant rate, the explicit temporal in Eqn.(5) is eliminated through the trivial transformation:
![]() (7)
(7)
Upon integration, Eqn.(7) gives
![]() (8)
(8)
If Ea/(RT) is replaced by x and integration limits transformed, Eqn.(8) becomes
![]() (9)
(9)
Eqn.(9) can be written as
![]() (10)
(10)
p(x) has no analytical solution but has many approximations[14-16], with one of the most popular being the Coats-Redfern method[11]. This method utilizes the asymptotic series expansion for approximating the exponential integral in Eqn.(10), giving
![]() (11)
(11)
Plotting the left hand side of Eqn.(11), which includes g(��) versus 1/T, gives Ea and A from the slope and intercept respectively. The model that gives the best linear fit is selected as the chosen model.
Despite the inability of this approach in kinetic analysis of non-isothermal process, many papers have been published based on this method in recent years [17-25] and conclusions based on this method continue to be published. In this study the reliability and accuracy of CR method to determine the kinetic model and kinetic parameters from non-isothermal data is evaluated using known simulated data.
3 Simulation
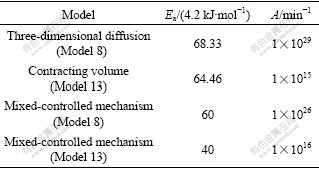
To study the reliability of CR method, three TGA curves were simulated. One curve was simulated using the contracting sphere (model 13), the second curve with three dimensional diffusion model (model 8, Janders Eqn.) and third curve with mixed control mechanism (models 8 and 13). In the mixed regime for the reaction extent less than 0.25, the reaction is chemical reaction controlled; at the reaction extent greater than 0.8, the reaction is controlled by three dimensional diffusion and at the reaction extent 0.25-0.8, both chemical reaction and diffusion are involved. Table 2 lists the kinetic parameters used for TGA curves simulation. These values of A and Ea are selected based on the experimental data reported in Refs.[23-25]. TGA curves were simulated at linear heating rates of 5, 7.5, 10 and 12.5 K/min. For all the curves, the initial temperature (T0) was considered to be 300 K. In the CR method a single TGA curve was used to determine kinetic parameters, so the data reported were extracted from TGA curves with heating rate of 10 K/min. Then by using the CR method, kinetic parameters were determined from simulated TGA curves and compared with original data.
Table 2 Values of A and Ea used for TGA curves simulation
4 Results and discussion
Simulated TGA curves for reactions with contracting sphere mechanism, three dimensional diffusion mechanism and mixed mechanism are shown in Figs.1-3 respectively. Also the initial temperature for all curves was considered to be same, but the reaction starts at different temperatures in simulated TGA curves. The range of reaction temperature for simulated TGA curves is in agreement with that of experimental works[23-25].
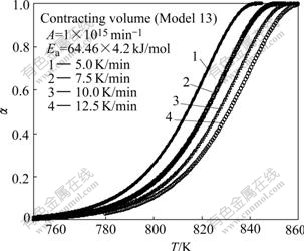
Fig.1 Simulated TGA curves for reaction with contracting sphere mechanism at different heating rates
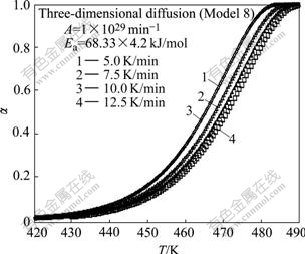
Fig.2 Simulated TGA curves for reaction with three- dimensional diffusion mechanism at different heating rates
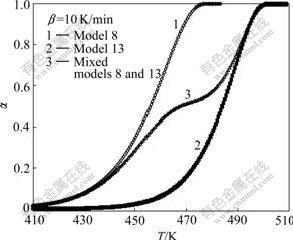
Fig.3 Simulated TGA curves for reaction with mixed-controlled mechanism at heating rate of 10 K/min
Fig.4 shows the plot of ln[g(��)/T2] versus 1/T for different models using values of a extracted from Fig.1. According to CR equation, if a correct model is selected for the reaction, the plot of ln[g(��)/T2] versus 1/T will be linear with high-correlation coefficient. Fig.4 reveals that all models show linear trend with correlation coefficient greater than 0.99. Table 3 lists the calculated kinetic parameters for different models with a from TGA simulated curves with contracting sphere mechanism by the CR method.
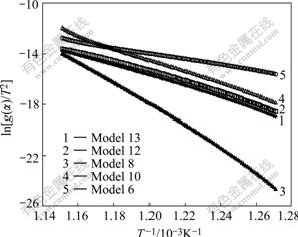
Fig.4 Plots of ln[g(��)/T2] versus 1/T for different models using values of �� from Fig.1
Table 3 Kinetic parameters extracted from Fig.4 using CR method
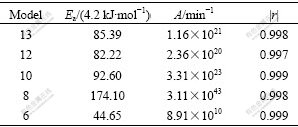
If model 13 in Table 3 is considered as reaction model, the activation energy calculated with CR method is 25% greater than real value and similarly pre-exponential factor (1.16��1021) is quite far from the assumed value (1��1015). These results show that the CR method reliability is not enough and cannot be used to kinetics assessment of reactions. The results of Fig.4 and Table 3 show that several chemical mechanisms (models 10, 12 and 13) are sufficiently similar in shape and calculated values of Ea and A are very close to each other. This indicates that calculated values of Ea are not directly proportional to reaction order because of the contribution from the term 2lnT in CR equation, which becomes relatively greater for the larger values of the reaction order[26].
Fig.5 shows the plot of ln[g(��)/T2] versus 1/T for different models using values of �� extracted from Fig.2.
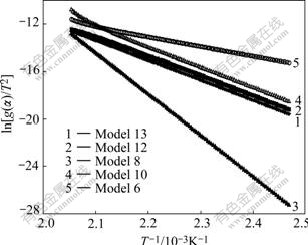
Fig.5 Plots of ln[g(��)/T2] versus 1/T for different models using values of �� extracted from Fig.2
Fig.5 shows all models have linear trend with correlation coefficient greater than 0.99. Table 4 lists the calculated kinetic parameters for different models with �� values from TGA simulated curve with three- dimensional diffusion mechanism by the CR method.
Table 4 Kinetic parameters extracted from Fig.5 using CR method
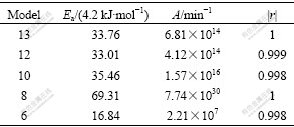
In this case if model 8 in Table 3 is considered as reaction model, the activation energy calculated with CR method is 1.4% greater than the real value and similarly pre-exponential factor (7.74��1030) is close to the assumed value (1��1029). Also the calculated values of A and Ea for three-dimensional diffusion mechanism are very close to the real values. However, in the first stage the correct model must be selected, which is impossible with CR method.
Comparison of Tables 3 and 4 shows that the magnitude of Ea calculated using diffusion model is nearly twice the values for chemical models (10, 12 and 13) and is ascribed to the dominant influence of characteristic diffusion exponent, n=0.5.
Fig.6 shows the plots of ln[g(��)/T2] versus 1/T for different models using the values of �� extracted from Fig.3.
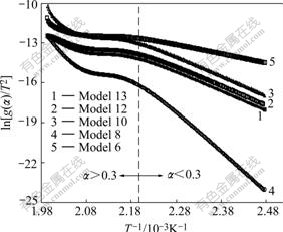
Fig.6 Plots of ln[g(��)/T2] versus 1/T for different models using values of �� extracted from Fig.3
It is clear from Fig.6 that for extents of reaction less than 0.3 that reaction is a single mechanism, the plot of ln[g(��)/T2] versus 1/T is linear. However, for extents of reaction greater than 0.3 that reaction has multi model mechanisms, the plot of ln[g(��)/T2] versus 1/T shows nonlinear trend. This means that the CR method can not be used to determine the reaction model. Therefore the kinetics of complex reactions, where the reaction model changes with the extent of reaction, cannot be analyzed with CR method. In most of solid state reactions (especially solid-gas reactions), a layer of products forms on the surface of un-reacted core. This layer may be porous or dense and this causes a change in the reaction mechanism by the increase in extent of reaction. Thus the CR method is ineffectual method in kinetic analysis of solid state reactions.
5 Conclusions
The CR method for the kinetic analysis of nonisothermal TGA data is shown to be unsuitable and inconsistencies exist in published kinetic results obtained using this approach. The results of this investigation show that for the reactions with simple mechanisms, the plot of ln[g(��)/T2] versus 1/T will be linear for all reaction kinetic models and the reaction mechanism can not be recognized with CR method. However, for reactions having complex mechanisms, the plot of ln[g(��)/T2] versus 1/T will not be linear and the CR method is not valid for kinetic analysis to find the reaction model and kinetic parameters.
References
[1] CONESA J A, MARCILLA A, CABALLERO J A, FONT R. Comments on the validity and utility of the different methods for kinetic analysis of thermogravimetric data [J]. J Anal Appl Pyrol, 2001, 58/59: 617-633.
[2] VYAZOVKIN S V, LESNIKOVICH A I. Error in determining activation energy caused by the wrong choice of process model [J]. Thermochim Acta, 1990, 165: 11-15.
[3] KOGA N, SESTAK J, MALEK J. Distortion of the Arrhenius parameters by the inappropriate kinetic model function [J]. Thermochim Acta, 1991, 188: 333-336.
[4] SUN T, ZHAO Y, JIN J, WANG D. J Therm Anal, 1995, 45: 1105-1109.
[5] HU Q P, CUI X G, YANG Z H. Studies on the non-isothermal kinetics of thermal decomposition of the mixed ligand complex [J]. J Therm Anal, 1997, 48: 1379-1384.
[6] CARP O, SEGAL E. Basic program to discriminate among mechanisms of solid-gas decomposition [J]. Thermochim Acta, 1991, 185: 111-118.
[7] VARHEGYI G, SAZAB? P, JAKAB E, TILL F. Least squares criteria for the kinetic evaluation of thermoanalytical experiments: Examples from a char reactivity study [J]. J Anal Appl Pyrol, 2001, 57: 203-222.
[8] MACIEJEWSKI M. Somewhere between fiction and reality [J]. J Therm Anal, 1992, 38: 51-70.
[9] CABALLERO J A, CONESA J A. Mathematical considerations for nonisothermal kinetics in thermal decomposition [J]. J Anal Appl Pyrolysis, 2005, 73: 85-100.
[10] GAO X, CHEN D, DOLLIMORE D. The correlation between the value of �� at the maximum reaction rate and the reaction mechanisms: A theoretical study [J]. Thermochim Acta, 1993, 223: 75-82.
[11] COATS A V, REDFERN J P. Kinetic parameters from thermogravimetric data [J]. Nature, 1964, 201: 68-69.
[12] VYAZOVKIN S, WIGHT C A. Model-free and model-fitting approaches to kinetic analysis of isothermal and nonisothermal data [J]. Thermochimica Acta, 1999, 340/341: 53-68.
[13] KHAWAM A, FLANGAN D R. Role of isoconversional methods in varying activation energies of solid state kinetics [J]. Thermochimica Acta, 2005, 436(1/2): 101-112.
[14] CHEN H J, LAI K M. Methods for determining the kinetics parameters from nonisothermal thermogravimetry [J]. J of Chem Eng of Japan, 2004, 37(9): 1172-1178.
[15] DOYLE C D. Kinetic analysis of thermogravimetric data [J]. J Appl Polym Sci, 1961, 5: 285.
[16] AGRAWAL R K, SIVASUBRAMANIAN M S. Integral approximations for nonisothermal kinetics [J]. AIChE J, 1987, 33: 1212-1214.
[17] AHMARUZZAMAN M, SHARMA D K. Non-isothermal kinetic studies on co-processing of vacuum residue, plastics, coal and petrocrop [J]. J Anal Appl Pyrolysis, 2005, 73: 263-275.
[18] P?REZ-MAQUEDA L A, S?NCHEZ-JIM?NEZ P E, CRIADO J M. Evaluation of the integral methods for the kinetic study of thermally stimulated processes in polymer science [J]. Polymer, 2005, 46: 2950-2954.
[19] BARRAL L, D?EZ F J, GARC?A-GARABAL S, L?PEZ J, MONTERO B, MONTES R, RAM?REZ C, RICO M. Thermodegradation kinetics of a hybrid inorganic-organic epoxy system [J]. European Polymer Journal, 2005, 41: 1662-1666.
[20] VACLAV S, SUSAK P. Pitch pyrolysis kinetics from single TG curve [J]. J Anal Appl Pyrolysis, 2004, 72: 249-252.
[21] VLAEV L T, MARKOVSKA I G, LYUBCHEV L A. Non-isothermal kinetics of pyrolysis of rice husk [J]. Thermochimica Acta, 2003, 406: 1-7.
[22] SRIKANTH S, CHAKRAVORTTY M. Non-isothermal thermoanalytical studies on the salt roasting of chalcopyrite using KCl [J]. Thermochimica Acta, 2001, 370: 141-148.
[23] DIEFALLAH E H M, GABAL M A, EL-BELLIHI A A, EISSA N A. Nonisothermal decomposition of CdC2O4-FeC2O4 mixtures in air [J]. Thermochimica Acta, 2001, 376: 43-50.
[24] JOSEPH K, SRIDHARAN R, GNANASEKARAN T. Kinetics of thermal decomposition of Th(C2O4)2��6H2O [J]. Journal of Nuclear Materials, 2000, 281: 129-139.
[25] HU H, CHEN Q, YIN Z, ZHANG P, ZOU J, CHE H. Study on the kinetics of thermal decomposition of mechanically activated pyrites [J]. Thermochimica Acta, 2002, 389: 79-83.
[26] GALWEY A K. Perennial problems and promising prospects in the kinetic analysis of nonisothermal rate data [J]. Thermochimica Acta, 2003, 407: 93-103.
Corresponding author: Reza Ebrahimi-Kahrizangi; E-mail: rezaebrahimi@iaun.ac.ir
