

J. Cent. South Univ. (2019) 26: 2328-2339
DOI: https://doi.org/10.1007/s11771-019-4177-5
Water leaching of arsenic trioxide from metallurgical dust with emphasis on its kinetics
YANG Kang(�)1, LIU Wei(��ά)1, ZHANG Tian-fu(������)1,YAO Li-wei(Ҧ��Ϊ)2, QIN Wen-qing(������)1
1. School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, China;
2. School of Metallurgy and Environment, Central South University, Changsha 410083, China
 Central South University Press and Springer-Verlag GmbH Germany, part of Springer Nature 2019
Central South University Press and Springer-Verlag GmbH Germany, part of Springer Nature 2019
Abstract:
Water leaching of As2O3 from metallurgical dust containing various metals was investigated, serving the purpose of dearsenization and simultaneous metal enrichment especially for indium. Effects of leaching temperature, liquid/solid ratio (LSR) and leaching time were studied. It was found that the initial dissolution was very fast but was then so inhibited by the increasingly dissolved As2O3, which makes it difficult to saturate enough arsenic in the leaching solution or in leaching out all the soluble arsenic with excess dosage of water within acceptable time (120 min). Only about 73% of As2O3 was extracted under the optimal conditions investigated. Two-step leaching showed similar trends and was thus unnecessary for improving As2O3 extraction. These observations could reasonably be accounted for the reversibility of the dissolution reaction. Kinetically, the leaching was described satisfactorily by the semi-empirical Avrami model with the apparent activation energy of 36.08 kJ/mol. The purity of the obtained product As2O3 could reach 98.7%, while the indium could be enriched in the leaching residue without loss.
Key words:
arsenic leaching; arsenic removal; arsenic trioxide; arsenate; arsenic recovery��
Cite this article as:
YANG Kang, LIU Wei, ZHANG Tian-fu, YAO Li-wei, QIN Wen-qing. Water leaching of arsenic trioxide from metallurgical dust with emphasis on its kinetics [J]. Journal of Central South University, 2019, 26(9): 2328-2339.
DOI:https://dx.doi.org/https://doi.org/10.1007/s11771-019-4177-51 Introduction
Arsenic is notorious for its toxicity which may lead to seriously environmental problems. It is well-documented that arsenic contamination has led to serious health risks such as cancer in lung, bladder, kidney and skin, as well as other skin changes in many countries all over the world [1]. In metallurgical field, arsenic is unwanted because it has been classified as one of the penalty elements, which has substantially reduced the economic value of the raw materials [2, 3]. Besides, its coexistence will complicate the process of recovering other metals of interest [4]. Therefore, arsenic removal from different ores/secondary resources prior to metal extraction has always been a topic for related researchers.
In the past decades, many efforts have been devoted to this subject. Due to the volatility of arsenic trioxide [5], roasting the arsenic-bearing material in appropriate vacuum environment has been proven to be an efficient means [4, 6-9]. However, secondary pollution which may be generated in this process is still a concern. Alternatively, hydrometallurgical direction has drawn attention as the route is relatively environment-friendly. In general, the reported work includes: 1) acid leaching [10], in which the useful metal may be dissolved into solution simultaneously with arsenic, suggesting that a following separation/purification step in aqueous phase is required; 2) alkaline leaching (especially in NaOH-Na2S media) [11-20], which has obviously drawn the most attention because of its good selectivity on arsenic; 3) pressure leaching [21, 22], which aims to further improve arsenic removal; 4) alkaline digestion followed by water leaching [23], which maximizes the alkaline efficiency.
The present paper deals with material containing arsenic as much as 48.5 wt%, which is the flue dust originated from the metallurgical process of lead-antimony ore. Despite of the high arsenic content, the presence of indium with a grade of about 0.3% is also confirmed in the dust, which turns out to be of great value, because over 90% of indium is recovered from by-product of zinc/lead metallurgy in China [24]. There are several side effects brought by arsenic during the recovery process of indium [25, 26]: 1) decreasing the indium recovery; 2) adversely affecting the quality of product sponge indium; 3) increasing the risk of producing extremely toxic gas AsH3. Hence it is necessary to remove arsenic from the dust as quickly as possible before it is subjected to indium recovery process.
On the other hand, given the fact that arsenic almost accounts for half of its weight, it may be a little arbitrary to treat this kind of dust as waste from metallurgy industry. After all, although arsenic is suffering from a shrinking market [27], it can still find some use in fields including manufacture of pesticides, wood preservatives, electronic components (i.e. semiconductors), glass, alloys, pigments and pharmaceuticals [6, 28]. LEIST et al [29] have also pointed out that, as literally quoted, where arsenic of a sufficient purity is produced (purity greater than 95%), the arsenic may be economically recovered for use primarily in the manufacture of the arsenical wood preservative, chromated copper arsenate (CCA), and ammonical copper-zinc arsenate (ACZA).
Therefore, we proposed that arsenic could be recovered as a by-product of interest. Instead of these aforementioned methods for arsenic extraction, water is chosen as a leaching agent in this study, because: 1) more than 85% of the arsenic in the dust exits as As2O3, which is soluble in water; 2) less impurities will be dissolved, which makes the following recovery step more convenient. Besides, it is surprisingly found that there were few details available on arsenic leaching behaviors by water, so it is of great meaning to carry out study on this subject.
2 Experimental
2.1 Material
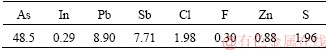
The dust was collected from a lead-antimony smelter located in Guangxi Province, China. Main compositions of the dust are shown in Table 1 (As, Pb, Sb and In are assayed by ICP; other composition are assayed by XRF).
Table 1 Main compositions of flue dust (mass fraction, %)
The particle size, analyzed in the absence of water by a laser particle size analyzer, is demonstrated in Figure 1. It is mostly distributed within the range from 0.224 ��m to 10 ��m, accounting for about 65% (V/V) of the dust. These fines tend to agglomerate easily, making it impossible to be screened without water. However, volume fraction because of the soluble As2O3 and the presence of water would be inappropriate. Therefore, effects of particle size will not be investigated.
The dust, as well as that leached by excess water several times to ensure that there was no more arsenic to be extracted by water, was subjected to XRD. The results are shown in Figure 2, indicating the presence of As2O3, Pb5(AsO4)3Cl, PbClF and Sb2O3 in the raw dust. Note that Pb5(AsO4)3Cl is not detectable in the raw dust probably due to its relatively low content, while PbClF has disappeared in the leached sample for unclear reason. Further analysis indicates that water soluble arsenic exists as As2O3, accounting for 88.72 wt% of the total arsenic content in the dust.
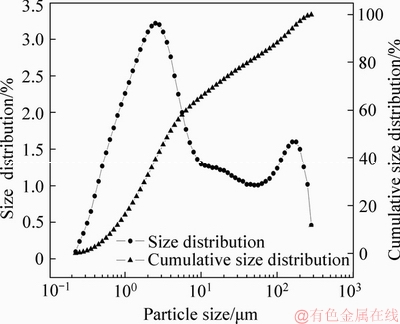
Figure 1 Particle size of dust
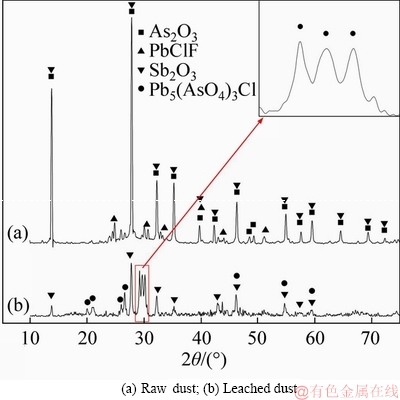
Figure 2 XRD results of dust and leached dust:
2.2 Leaching procedure
The leaching procedure was performed in a three-neck flask (500 mL) fixed in a water bath. Tap water (pH=8.02, 30 ��C) was used as leaching agent because it was of more practical importance. The dust was weighed and introduced into the flask after the prepared leaching solution had reached the expected temperature. The pH value of the leaching system was continually monitored at 30 ��C, 40 ��C and 60 ��C (higher temperature would spoil the probe of the pH measurer). All the leaching processes had been proceeded at a stirring velocity of 300 r/min. The effects of stirring rate were eliminated because the results were identical with stirring rate ranging from 200 to 500 r/min at 95 ��C, which was unnecessary to report graphically in the following sectors.
A syringe was used to draw out the slurry (5-10 mL) from the reactor at regulated intervals, and then the particles were immediately separated from the aqueous phase with a syringe filter with porosity of 0.45 ��m. The obtained solution was quickly diluted and sealed in a centrifuge tube, and was subsequently sent to ICP for analysis of arsenic content. Leaching solution of each run of the experiments was collected and was then partially subjected to evaporation to obtain the product As2O3.
The experiments were performed twice and the results of arsenic concentration from the two runs were correspondingly averaged for the calculation of arsenic extraction efficiency, which was evaluated by percentage of arsenic dissolved with respect to the As2O3 content:
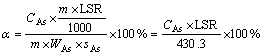 (1)
(1)
where �� is the relative leaching efficiency of arsenic; CAs is the mean arsenic concentration in the leaching solution from the two runs (calculated as elemental As), g/L; LSR is the liquid/solid ratio, mL/g; m is the mass of the sample, g; WAs is the grade of arsenic in the dust, which is 0.4850; and sAs is the content of arsenic in the form of As2O3 with respect to the total arsenic, which is 0.8872.
3 Results and discussion
3.1 Water leaching
As presented by POKROVSKI et al [30], dissolution of As2O3 is very sensitive to temperature (Figure 3). Figure 3 is originally presented in a table in the form of lgwAs (mol As/kg water). The uncertainties (dots with error range) are given for the 95% confidence level. The solubility of As2O3 is about 11 g/L (calculated as element As) at 22 ��C, and is increased to more than 60 g/L at 98 ��C. Regardless of a relatively low solubility, this provides the feasibility of the arsenic recovery directly by water.
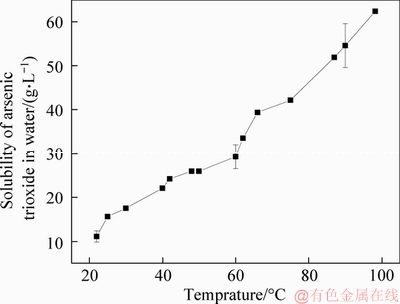
Figure 3 Dependence of As2O3 solubility on temperature [30]
Figure 4 shows the effects of leaching time, leaching temperature and LSR on the arsenic extraction. Note that LSR actually represents the water dosage.
As shown in Figure 4(a), raising temperature was obviously favorable for arsenic extraction. Only 14.58% of arsenic was removed while the dust was leached out at 30 ��C for 120 min, and 72.97% of arsenic was dissolved into the water when the temperature was elevated to 95 ��C. Meanwhile, more water was also, as expected and shown in Figure 4(b), an efficient means to generate better arsenic removal. The sample leached at 95 ��C for 120 min was decreased to around 30% once LSR dropped to 3 mL/g.
On the other hand, it was necessary to present results of CAs corresponding to Figure 4, as demonstrated in Figure 5, because it was surprisingly found that none of these conducted runs had reached saturation within 120 min, leaving enough arsenic in the dust. The highest CAs (about 45 g/L) was obtained by leaching at 95 ��C for 120 min with LSR=3 mL/g (Figure 5(b)). Similarly, it failed to leach all the arsenic out within 120 min regardless of the excess dosage of water.
Generally, results of Figures 4 and 5 have actually suggested that the arsenic leaching was so time-dependent that it could easily be differentiated into two stages: 1) a quick extraction of arsenic within the initial 10 min (Stage I); 2) the following comparably slow dissolution (Stage II). The consequence was that, compared with Stage I, Stage II took more time only to generate a much less arsenic extraction. This led to the difficulty in saturating the leaching solution and in leaching out all the soluble arsenic with excess dosage of water within acceptable leaching time of 120 min.
From a technical point of view, this observation was of importance because it implied a limit on CAs where the leaching started to become so slow that reusing of leaching solution in order to get saturated would be inefficient. One possible explanation for this phenomenon would be discussed later.
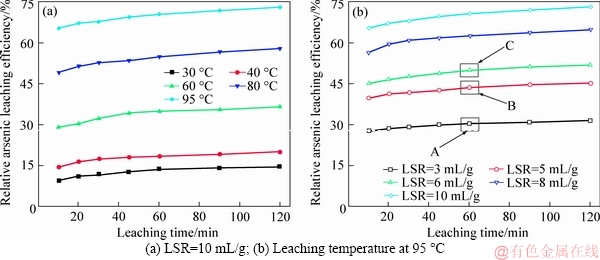
Figure 4 Changes of arsenic extraction during leaching:
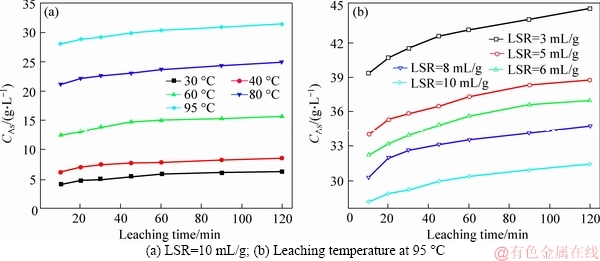
Figure 5 Changes of CAs during leaching:
Further experiments of leaching were then conducted to extract arsenic from the leached residue. The purposes of these runs were: 1) to find out whether the leaching behaviors of the remaining As2O3 would inherit the trend of Stage II of the previous leaching step; and 2) to evaluate the feasibility of optimizing As2O3 extraction by two- step leaching.
The residues subjected to another leaching process are marked in Figure 4(b) with capitals A, B and C. Before the leaching, the residues were dried at 80 ��C for 24 h. All the experiments were performed at 95 ��C. The results are showed in Figure 6.
First of all, it was found that whichever the sample was, observation similar to the previous leaching step was made that As2O3 extraction endured a process consisting of Stage I and Stage II. This indicated that whatever caused the changes of arsenic leaching behavior, it was not eliminated by the previous leaching step. In the meantime, it was because in the presence of Stage I that, for samples leached with total LSR=8 mL/g in Figure 6(a) and LSR=10 mL/g in Figure 6(b), the cumulative As2O3 leaching efficiency had reached about 65% and 73% within 10 min (70 min in total), respectively. Whereas in one-step leaching process at similar LSR, it took 120 min for the same amount of arsenic to be extracted. However, this benefit could easily be counteracted economically by the necessary involvement of another solid-liquid separation process. In addition, it could be seen that even when the total LSR was elevated to larger than 10 mL/g, more than 10% of As2O3 was still left in the residue after being leached for 90 min (Figures 6(b) and (c)).
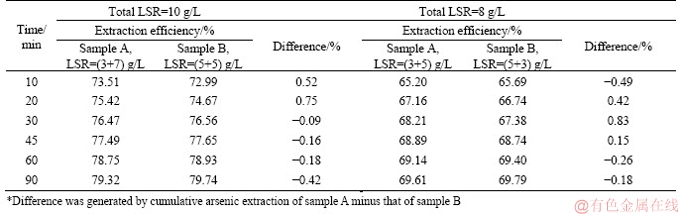
Secondly, there seemed to be no apparent difference between different samples leached with the same total LSR and duration in the two-step leaching process. In another words, as long as the total LSR was equal, whatever was the LSR for the previous/current leaching step, As2O3 extraction with the same leaching time would be cumulatively similar. Comparison was made as shown in Table 2 for a better description.
As seen in Table 2, all the differences on cumulative As2O3 extraction between different samples are less than 1%, which obviously could be explained by the experimental errors. This was an important phenomenon as it rejected the possibility of optimizing arsenic extraction by exchanging the sequence of LSR.
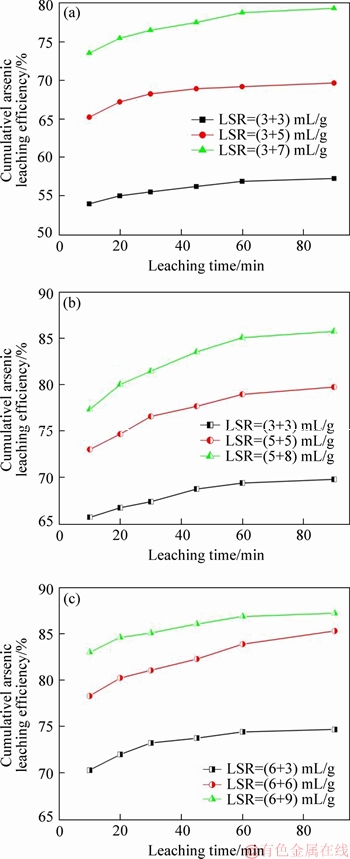
Figure 6 Effects of LRS and leaching time on cumulative arsenic extraction (*LSR=(X+Y) mL/g means that the sample is previously leached with LSR=X mL/g and currently leached with LSR=Y mL/g with respect to the raw dust, the total LSR being (X+Y) mL/g)
It should be conclusively seen that two-step leaching was not a necessary option for optimization, because it had limited improvement for the extraction at the cost of additional phase separation procedure.
Table 2 Comparison of As2O3 extraction efficiency between different runs with same total LSR
3.2 Leaching chemistry of dust
3.2.1 Dissolution chemistry
During leaching, As2O3 is ready to be dissolved by water through reaction [30]:
 (2)
(2)
The formed H3AsO3 (also written as As(OH)3) is weak acid and is able to deprotonate via reactions below:
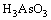

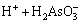 (3)
(3)

 (4)
(4)
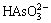
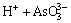 (5)
(5)
With the first pKa of arsenite at 9.23, the neutral species will dominate in most natural aqueous solutions [31], and with the other two pKa values (pK2=12.13, pK3=13.40), it is easy to calculate the distribution of As(III) species with respect to the change of pH value (Figure 7). It can be noted that the deprotonated anions have not obviously shown up until the pH value has reached 8.0. This calculation is in agreement with the experimental Raman spectroscopic results of GOUT et al [32], who have also pointed out that spectrum is not significantly affected by the temperature (from 20 up to 275 ��C) and further concluded that the pyramidal As(OH)3 molecule is predominant at all temperatures investigated in these solutions (wAs��0.5 mol/kg, that is, approximately, CAs��37.5 g/L).
In order to find out the species of arsenic in the leaching solution, the change of pH was traced for the leaching system at temperature 30, 40 and 60 ��C. It was observed that the pH value was quickly decreased from 8.02, 7.97 and 7.84 to 3.58, 3.45 and 3.06 within 30 s, respectively. Apparently, the dissolution of As2O3 and the following deprotonation as indicated by Eqs. (2)-(5) had contributed mainly to the instant drop of pH value. With the increase on time, the pH value fluctuated slightly within the range of ��0.08. The change was so irregular that it was hard to tell whether it was caused by the instrumental error or originated from the leaching system. Nevertheless, this was not important because the observation had suggested that, since there was no alkali complex in the residue enough to raise the pH value to basic system, H3AsO3 must be the predominant species in the solution.
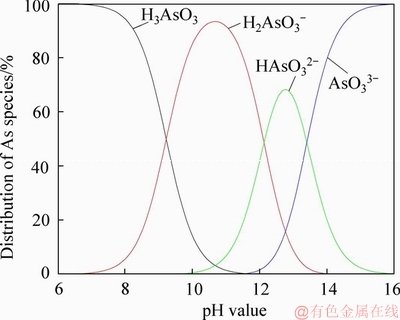
Figure 7 Dependence of As(III) species on pH value at 25 ��C (pK1=9.23; pK2=12.13; pK3=13.40)
It was then reasonable to try to connect the arsenic extraction behaviors with the drop of pH value in the leaching system, but experiments in which the dust was leached at 95 ��C by acidic solution with different pH values (pH=2, 3, 4, adjusted using H2SO4) had ruled out this possibility, because the difference between these runs and those leached by fresh water was barely enough to explain the change of leaching behavior of arsenic. As a matter of fact, the decrease on pH value of the leaching agent had hardly changed the pattern of the leaching process, of which details were thus unnecessary to be reported here.
Inspired by the work reported by FRANKE et al [33], in which effects of dissolved aluminum (target element in the research) on the dissolution process were examined, an idea came up that arsenic extraction might be affected by the concentration of itself in the leaching solution. Accordingly, another set of experiments were carried out to verify it.
The raw dust was leached at 95 ��C for 60 min with LSR=10 mL/g employing prepared solution varying in As2O3 content. The results are presented in Table 3.
Table 3 Effects of dissolved As2O3 on dissolution (g/L)
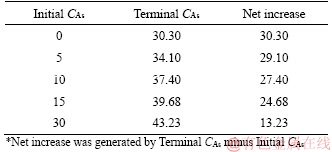
If the leaching process was independent on the dissolved arsenic, the net increases on CAs of different samples should be identical (or close with acceptable errors) to each other. But as expected, it was evidently decreased with increasing initial CAs. The inhibition was slight at initial CAs=5 g/L, but significant when the initial CAs was increased to 30 g/L. Therefore, the As2O3 extraction was affected by the dissolved As2O3 itself. The self-inhibition could explain well the change of As2O3 extraction behaviors during the leaching process, and one possible explanation for this observation was that Eq. (2) was reversible.
Following Eq. (2), it was obvious that the arsenic leaching rate was determined by both the formation rate and decomposition rate of H3AsO3. In the beginning, the dissolution rate of As2O3 was rapid and H3AsO3 was quickly formed. In the meantime, the formed H3AsO3 started to decompose into As2O3 and H2O, but since CAs was low, the rate was ignorable. However, when more and more arsenic was dissolved into the water, the backward reaction rate was increased with the increase of CAs, resulting in the significant decrease on the net leaching rate. Therefore, the occurrence of the reverse reaction should be the reason for the difference of arsenic leaching behavior between Stage I and Stage II. Moreover, this could also explain the observation from two-step leaching process. As a matter of fact, it was obvious that the previous leaching could not change the nature of the dissolution reaction, and thus arsenic behavior in the second leaching step had to exhibit the same trend.
3.2.2 Morphological changes during dissolution
Different samples were subjected to scanning electron microscope (SEM) to reveal more details on the microscopic change of the dust particles during the leaching process, as shown in Figure 8. As could be clearly seen, crystal particles and amorphous phases were both observable in the raw dust (Figure 8(a) for sample a) as well as in the leached dusts (Figures 8(b) and (c) for sample b and sample c). Generally, crystal structure of sample a was relatively well presented, while some crystals of sample b and sample c appeared to be either rough or damaged. It was unlikely that these were caused without exogenous force. After the dust was leached by excess water, there were no crystals found in observable size in the residue (Figure 8(d) for sample d). These observations had strongly suggested that the changes on the crystal structure and the disappearance of observable crystals had represented the As2O3 dissolution process.
3.3 Kinetics discussion
Attempts were failed when fitting data presented above into any shrinking core model, which was usually used to describe many metallurgic processes. Instead, it was satisfactorily characterized by the semi-empirical Avrami model (Eq. (6)), which had also been successfully adopted to explain various metal extractions [33-39].
The Avrami model is arranged here in the form of natural logarithm as follow:
ln(-ln(1-x))=lnk+nlnt (6)
where x=�� is the fraction of As2O3 dissolved; k is the overall leaching rate constant, min-n; t is the leaching time, min; and n is the Avrami index.
As shown in Figure 9, the plot of ln(-ln(1-x)) versus lnt under different temperatures was made by statistical means of linear regression using data from Figure 4(a). Accordingly, the values of the parameters including n, lnk and correlation coefficient (R2) are given in Table 4. The results confirmed with satisfactory R2 the linear relationship between ln(-ln(1-x)) and lnt at each temperature investigated.

Figure 8 SEM images of different samples:
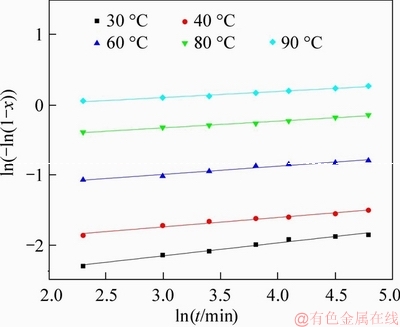
Figure 9 Plots of ln(-ln(1-x)) vs lnt under different temperatures
Table 4 Values of n, lnk and correlation coefficients R2
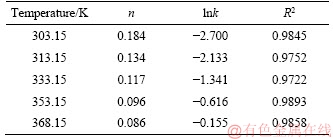
According to KABAI [39], the characteristic of the leaching process could be specified by n value : 1) for n<1, the rate was initially infinite and it continually decreased with increasing time; 2) for n=1, the initial rate was finite; and 3) for n>1, the leaching would exhibit an initial rate approaching zero.
Therefore, since all the values of n appeared to be much less than one (Table 4), an infinite dissolution rate at the beginning followed by a finite one was expectable. This kinetically explained the mentioned difference between As2O3 leaching behaviors in Stage I and that in Stage II. The kinetic data are then plotted in Figure 10 with natural logarithm of rate constant versus 1/T according to Arrhenius equation, by which the value of the apparent activation energy was calculated to be 36.08 kJ/mol with fairly satisfactory linear relationship (R2=0.9990).
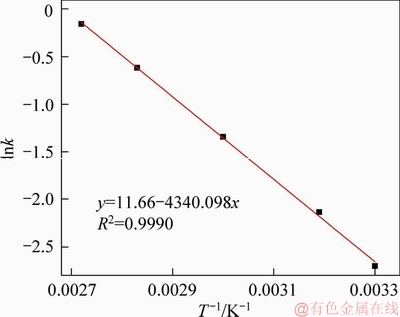
Figure 10 Arrhenius plot for As2O3 leaching process
On the other hand, KABAI [39] had also pointed out that the value of n in Eq. (6) was independent on process conditions but was a function of property and geometry of the nucleated phase. This suggested that the value of n would remain the same (with explainable/acceptable error) whatever changes were made on experiment conditions. Many other reports had confirmed this conclusion [33, 37, 38], while some n values reported were, more or less, evidently fluctuated [34, 35, 40, 41].
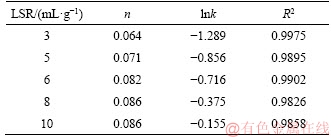
In the present paper, value of n seemed to be sensitive to the change of temperature (as had already been discussed and shown in Table 4), but varying LSR had less effects on it (shown in Figure 11 and Table 5). The reason was yet to be found, but it was highly possible that this phenomenon was related to the reversibility of the dissolution reaction as indicated by Eq. (2).
3.4 Product analysis
The content of the As2O3 obtained tentatively by evaporation of the collected leaching solution was determined to be 98.7%, indicating that As2O3 recovery with satisfactory purity from the leaching solution was feasible. The XRD result shown in Figure 12 confirms that the crystal structure of the generated product was well formed.
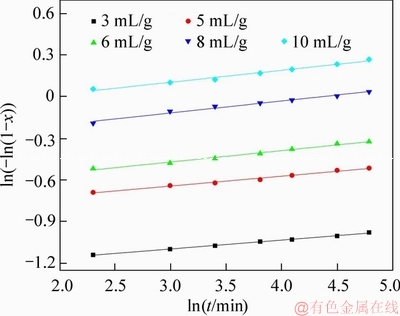
Figure 11 Plots of ln(-ln(1-x)) vs lnt under different LSRs
Table 5 Values of n, lnk and correlation coefficients R2
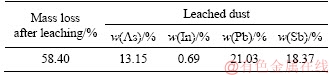
It should also be mentioned that indium, lead and antimony were not leached out. Concentrations of indium in the collected and mixed leaching solution (CAs=15.89 g/L) were much less than 1 mg/L, while these of the other two were lower than 100 mg/L. Besides, quantitative analysis of the residue leached by excess water had confirmed that the involvement of all the three elements during the As2O3 extraction was actually ignorable, as shown in Table 6.
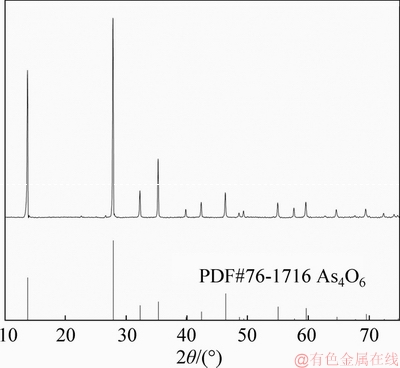
Figure 12 XRD result of product
Table 6 Elements content of interests in leached dust
4 Conclusions
The present paper investigated the water leaching of As2O3 from metallurgical dust containing indium. Based on results and discussion above, conclusions are drawn as follows:
1) As2O3 in the dust was dissolved by water through the following reaction:
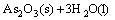
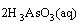
and the reversibility of this reaction was the reason for the subsequent inhibition during the extraction.
2) About 73% of As2O3 was extracted while the dust was leached for 120 min at 95 ��C with LSR=10 mL/g. It was not necessary to take a two-step leaching process for better extraction because the improvement was limited due to the occurrence of the reversible dissolution reaction.
3) Kinetically, the leaching could be described satisfactorily by the semi-empirical Avrami model with the activation energy being 36.08 kJ/mol.
4) The purity of the product As2O3 could reach 98.7%, and simultaneously, all the indium was left in the residue and was enriched. If the whole arsenic was removed, indium content would be increased from 0.29% in the raw dust to 0.69% in the leached residue.
References
[1] HAN C, LI H, PU H, YU H, DENG L, HUANG S, LUO Y. Synthesis and characterization of mesoporous alumina and their performances for removing arsenic (V) [J]. Chemical Engineering Journal, 2013, 217: 1-9. DOI: 10.1016/j.cej. 2012.11.087.
[2] LONG G, PENG Y, BRADSHAW D. A review of copper�Carsenic mineral removal from copper concentrates [J]. Minerals Engineering, 2012, 36�C38: 179-186. DOI: 10.1016/ j.mineng.2012.03.032.
[3] MIHAJLOVIC I, STRBAC N, ZIVKOVIC Z, KOVACEVIC R, STEHERNIK M. A potential method for arsenic removal from copper concentrates [J]. Minerals Engineering, 2007, 20(1): 26-33. DOI: 10.1016/j.mineng.2006. 04.006.
[4] LIN D, QIU K. Removing arsenic from anode slime by vacuum dynamic evaporation and vacuum dynamic flash reduction [J]. Vacuum, 2012, 86(8): 1155-1160. DOI: 10.1016/j.vacuum.2011.10.023.
[5] SAFARZADEH M S, MILLER J D, HUANG H H. The behavior of arsenic trioxide in non-ferrous extractive metallurgical processing [J]. Metallurgical Research and Technology, 2014, 111(2): 95-105. DOI: 10.1051/metal/ 2014020.
[6] LIN D, QIU K. Removal of arsenic and antimony from anode slime by vacuum dynamic flash reduction [J]. Environmental Science and Technology, 2011, 45(8): 3361-3366. DOI: 10.1021/es103424u.
[7] YIN Z, LU W, XIAO H. Arsenic removal from copper�Csilver ore by roasting in vacuum [J]. Vacuum, 2014, 101: 350-353. DOI: 10.1016/j.vacuum.2013.10.005.
[8] SHIBAYAMA A, TAKASAKI Y, WILLIAM T, YAMATODANI A, HIGUCHI Y, SUNAGAWA S, ONO E. Treatment of smelting residue for arsenic removal and recovery of copper using pyro-hydrometallurgical process [J]. Journal of Hazardous Materials, 2010, 181(1-3): 1016-1023. DOI: 10.1016/j.jhazmat.2010.05.116.
[9] PETERSON M, TWIDWELL L G. Removal of arsenic from lead smelter speiss [J]. Journal of Hazardous Materials, 1985, 12(3): 225-229. DOI: 10.1016/0304-3894(85) 85009-3.
[10] KE J, QIU R, CHEN C. Recovery of metal values from copper smelter flue dust [J]. Hydrometallurgy, 1984, 12(2): 217-224. DOI: 10.1016/0304-386X(84)90035-5.
[11] LI Y, LIU Z, LI Q, ZHAO Z, LIU Z, ZENG L. Removal of arsenic from Waelz zinc oxide using a mixed NaOH-Na2S leach [J]. Hydrometallurgy, 2011, 108(3, 4): 165-170. DOI: 10.1016/j.hydromet.2011.04.002.
[12] LI Y, LIU Z, LI Q, ZHAO Z, LIU Z, ZENG L, LI L. Removal of arsenic from arsenate complex contained in secondary zinc oxide [J]. Hydrometallurgy, 2011, 109(3, 4): 237-244. DOI: 10.1016/j.hydromet.2011.07.007.
[13] TONGAMP W, TAKASAKI Y, SHIBAYAMA A. Arsenic removal from copper ores and concentrates through alkaline leaching in NaHS media [J]. Hydrometallurgy, 2009, 98(3, 4): 213-218. DOI: 10.1016/j.hydromet.2009.04.020.
[14] AWE S A, SANDSTR M  . Selective leaching of arsenic and antimony from a tetrahedrite rich complex sulphide concentrate using alkaline sulphide solution [J]. Minerals Engineering, 2010, 23(15): 1227-1236. DOI: 10.1016/ j.mineng.2010.08.018.
. Selective leaching of arsenic and antimony from a tetrahedrite rich complex sulphide concentrate using alkaline sulphide solution [J]. Minerals Engineering, 2010, 23(15): 1227-1236. DOI: 10.1016/ j.mineng.2010.08.018.
[15] BAL  P, ACHIMOVI
P, ACHIMOVI OV M. Selective leaching of antimony and arsenic from mechanically activated tetrahedrite, jamesonite and enargite [J]. International Journal of Mineral Processing, 2006, 81(1): 44-50. DOI: 10.1016/j.minpro.2006.06.004.
OV M. Selective leaching of antimony and arsenic from mechanically activated tetrahedrite, jamesonite and enargite [J]. International Journal of Mineral Processing, 2006, 81(1): 44-50. DOI: 10.1016/j.minpro.2006.06.004.
[16] FERN NDEZ M A, SEGARRA M, ESPIELL F. Selective leaching of arsenic and antimony contained in the anode slimes from copper refining [J]. Hydrometallurgy, 1996, 41(2, 3): 255-267. DOI: 10.1016/0304-386X(95)00061-K.
[17] TONGAMP W, TAKASAKI Y, SHIBAYAMA A. Selective leaching of arsenic from enargite in NaHS-NaOH media [J]. Hydrometallurgy, 2010, 101(1, 2): 64-68. DOI: 10.1016/ j.hydromet.2009.11.020.
[18] VIR KOVA E, FEDOR J. Leaching behaviour of arsenic trisulphide with sodium hydroxide solution [J]. Hydrometallurgy, 1991, 27(1): 1-6. DOI: 10.1016/0304- 386X(91)90073-U.
[19] GUO X, YI Y, SHI J, TIAN Q. Leaching behavior of metals from high-arsenic dust by NaOH-Na2S alkaline leaching [J]. Transactions of Nonferrous Metals Society of China, 2016, 26(2): 575-580. DOI: 10.1016/S1003-6326(16) 64118-3.
[20] DELFINI M, FERRINI M, MANNI A, MASSACCI P, PIGA L. Arsenic leaching by Na2S to decontaminate tailings coming from colemanite processing [J]. Minerals Engineering, 2003, 16(1): 45-50. DOI: 10.1016/S0892-6875 (02)00262-5.
[21] LI Y, LIU Z, LI Q, LIU F, LIU Z. Alkaline oxidative pressure leaching of arsenic and antimony bearing dusts [J]. Hydrometallurgy, 2016, 166: 41-47. DOI: 10.1016/ j.hydromet.2016.07.010.
[22] PADILLA R, RODR GUEZ G, RUIZ M C. Copper and arsenic dissolution from chalcopyrite �C enargite concentrate by sulfidation and pressure leaching in H2SO4-O2 [J]. Hydrometallurgy, 2010, 100(3, 4): 152-156. DOI: 10.1016/ j.hydromet.2009.11.006.
[23] RUIZ M C, GRANDON L, PADILLA R. Selective arsenic removal from enargite by alkaline digestion and water leaching [J]. Hydrometallurgy, 2014, 150: 20-26. DOI: 10.1016/j.hydromet.2014.09.004.
[24] CAO Y. Recovery in from as dust of copper smelter process [J]. Hunan Nonferrous Metals, 2005, 21(1): 5-8. http://en.cnki.com.cn/Article_en/CJFDTotal-HNYJ200501003.htm. (in Chinese)
[25] CHEN W, NIU Q, WANG Y, ZENG G, LI Y, PENG Y, HUANG J. Study on technology of eliminating arsenic in process of producing indium [J]. Journal of Hunan University (Natural Sciences), 2001, 28(5): 96-99. http://en.cnki.com.cn/Article_en/CJFDTotal-HNDX200105018.htm. (in Chinese)
[26] WEI Y. Discussion and practice of arsenic removal process for the recovering indium in hydrometallurgy zinc [J]. Nonferrous Mining and Metallurgy, 2015, 31(3): 33-36. http://www.en.cnki.com.cn/Article_en/CJFDTotal-YSKY201503010.htm. (in Chinese)
[27] YANG K, QIN W, LIU W. Extraction of metal arsenic from waste sodium arsenate by roasting with charcoal powder [J]. Metals, 2018, 8(7): 542. DOI: 10.3390/met8070542.
[28] WILSON S C, LOCKWOOD P V, ASHLEY P M, TIGHE M. The chemistry and behaviour of antimony in the soil environment with comparisons to arsenic: A critical review [J]. Environmental Pollution, 2010, 158(5): 1169-1181. DOI: 10.1016/j.envpol.2009.10.045.
[29] LEIST M, CASEY R J, CARIDI D. The management of arsenic wastes: problems and prospects [J]. Journal of Hazardous Materials, 2000, 76(1): 125-138. DOI: 10.1016/ S0304-3894(00)00188-6.
[30] POKROVSKI G, GOUT R, SCHOTT J, ZOTOV A, HARRICHOURY J C. Thermodynamic properties and stoichiometry of As(III) hydroxide complexes at hydrothermal conditions [J]. Geochimica et Cosmochimica Acta, 1996, 60(5): 737-749. DOI: 10.1016/0016-7037(95)00 427-0.
[31] O'DAY P. Chemistry and mineralogy of arsenic [J]. Elements, 2006, 2(2): 77-83. DOI: 10.2113/gselements.2.2.77.
[32] GOUT R, POKROVSKI G, SCHOTT J, ZWICK A. Raman spectroscopic study of arsenic speciation in aqueous solutions up to 275 ��C [J]. Journal of Raman Spectroscopy, 1998, 28(9): 725-730. DOI: 10.1002/(SICI)1097-4555 (199709)28:9<725::AID-JRS169>3.0.CO;2-9.
[33] FRANKE M D, ERNST W R, MYERSON A S. Kinetics of dissolution of alumina in acidic solution [J]. AIChE Journal, 1987, 33(2): 267-273. DOI: 10.1002/aic.690330213.
[34] HE G, ZHAO Z, WANG X, LI J, CHEN X, HE L, LIU X. Leaching kinetics of scheelite in hydrochloric acid solution containing hydrogen peroxide as complexing agent [J]. Hydrometallurgy, 2014, 144-145(4): 140-147. DOI: 10. 1016/j.hydromet.2014.02.006.
[35] ZHENG Y J, CHEN K K. Leaching kinetics of selenium from selenium�Ctellurium-rich materials in sodium sulfite solutions [J]. Transactions of Nonferrous Metals Society of China, 2014, 24(2): 536-543. DOI: 10.1016/S1003- 6326(14)63093-4.
[36] LI G, RAO M, JIANG T, HUANG Q, PENG Z. Leaching of limonitic laterite ore by acidic thiosulfate solution [J]. Minerals Engineering, 2011, 24(8): 859-863. DOI: 10.1016/ j.mineng.2011.03.010.
[37] KANUNGO S B, JENA P K. Studies on the dissolution of metal values in manganese nodules of Indian Ocean origin in dilute hydrochloric acid [J]. Hydrometallurgy, 1988, 21(1): 23-39. DOI: 10.1016/0304-386X(88)90014-X.
[38] KESKINLER B, BAYRAMO LU M. Kinetics of phosphoric acid production by decomposition of monocalcium phosphate in ethanol [J]. International Journal of Mineral Processing, 1992, 36(3): 259-271. DOI: 10.1016/ 0301-7516(92)90048-2.
LU M. Kinetics of phosphoric acid production by decomposition of monocalcium phosphate in ethanol [J]. International Journal of Mineral Processing, 1992, 36(3): 259-271. DOI: 10.1016/ 0301-7516(92)90048-2.
[39] KABAI J. Determination of specific activation energies of metal oxides and metal oxide hydrates by measurement of the rate of dissolution [J]. Acta Chim Acad Scientiarium Hungaricae, 1973, 78: 57-73.
[40] ZHANG R, MA S, YANG Q, ZHENG S, ZHANG Y, KIM N, HONG S. Research on NaCaHSiO4 decomposition in sodium hydroxide solution [J]. Hydrometallurgy, 2011, 108 (3, 4): 205-213. DOI: 10.1016/j.hydromet.2011.04.010.
[41] HULBERT S F, HUFF D E. Kinetics of alumina removal from a calcined kaolin with nitric, sulfuric, and hydrochloric acids [J]. Clay Minerals, 1970, 8(3): 337-345. DOI: 10.1180/claymin.1970.008.3.11.
(Edited by ZHENG Yu-tong)
���ĵ���
��ұ���̳���ˮ����ȡAs2O3�Ķ���ѧ�о�
ժҪ�����IJ�����ˮ�������Ӻ��ж��ֽ�����ұ���̳�����ȡAs2O3��Ŀ�����������ͬʱʹ�����м۽����ر��������Ը�������Ҫ�о��˽����¶ȡ�����Һ�̱�(LSR)�ͽ���ʱ���������̵�Ӱ�졣�����ʾ���ڽ����������ܽ⼫�죬�����Ž���Һ����Ũ�ȵ���ߣ���Ľ����ܵ����ơ���ʹ�ý���Һ���Դﵽ���ͣ���ʹʹ�ù�����ˮҲ�����ڿɽ��ܵ�ʱ�䷶Χ��(120 min)����ȫ�������������ʵ�������£�As2O3�Ľ����ʽ�Ϊ73%��������������������������Ч���������Եĸ��ơ���Щ�������As2O3�ܽⷴӦ�Ŀ����Ե��Խ��͡�����ѧ�ϣ��������̿��ð뾭���Avramiģ�ͽ��������������㣬��Ӧ���Ϊ36.08 kJ/mol��ʵ������As2O3���ȿɴ�98.7%����ˮ������������δ������ʧ��
�ؼ��ʣ���Ľ��������飻As2O3�������Σ������
Foundation item: Project(51874356) supported by the National Natural Science Foundation of China
Received date: 2018-01-14; Accepted date: 2018-11-08
Corresponding author: QIN Wen-qing, PhD, Professor; Tel: +86-13508473871; E-mail: qinwenqing2@126.com; ORCID: 0000-0001- 6290-7036
Abstract: Water leaching of As2O3 from metallurgical dust containing various metals was investigated, serving the purpose of dearsenization and simultaneous metal enrichment especially for indium. Effects of leaching temperature, liquid/solid ratio (LSR) and leaching time were studied. It was found that the initial dissolution was very fast but was then so inhibited by the increasingly dissolved As2O3, which makes it difficult to saturate enough arsenic in the leaching solution or in leaching out all the soluble arsenic with excess dosage of water within acceptable time (120 min). Only about 73% of As2O3 was extracted under the optimal conditions investigated. Two-step leaching showed similar trends and was thus unnecessary for improving As2O3 extraction. These observations could reasonably be accounted for the reversibility of the dissolution reaction. Kinetically, the leaching was described satisfactorily by the semi-empirical Avrami model with the apparent activation energy of 36.08 kJ/mol. The purity of the obtained product As2O3 could reach 98.7%, while the indium could be enriched in the leaching residue without loss.
