
Trans. Nonferrous Met. Soc. China 23(2013) 2700-2707
First principles study of structural, electronic and mechanical properties of transition metal hydrides (TMH, TM=Mo,Tc, Ru)
G. SUDHA PRIYANGA1, A. T. ASVINI MEENAATCI1, R. RAJESWARA PALANICHAMY1, K. IYAKUTTI2
1. N.M.S.S. Vellaichamy Nadar College, Madurai, Tamil nadu-625019, India;
2. Department of Physics and Nanotechnology, SRM University, Chennai, Tamil nadu-603203, India
Received 1 November 2012; accepted 7 April 2013
Abstract:
The structural, electronic and mechanical properties of transition metal hydrides (TMH, TM=Mo, Tc, Ru) are investigated by means of first principles calculation based on density functional theory with generalized gradient approximation. Among the five crystallographic structures that have been investigated, the cubic phase is found to be more stable than the hexagonal ones. A structural phase transition from ZB to WC in MoH, NaCl to NiAs in TcH and NaCl to ZB to NiAs in RuH is also predicted under high pressure. The calculated elastic constants indicate that all the three hydrides are mechanically stable at ambient pressure.
Key words:
ab-initio method; structural phase transition; electronic properties; elastic property;
1 Introduction
During the past years, the interaction between transition metals and H2 has raised great interest among the researchers, as transition metals are not only regarded as promising potential materials for H2 storage [1], but also used as structural materials in nuclear reactors due to their low neutron absorption cross section, high mechanical strength and excellent corrosion resistance at elevated temperatures [2]. Accurate first principles methods can complement and help to interpret high pressure experiments, which can provide a detailed description of the structural and bonding changes that a material undergoes under extreme conditions. The behavior of H2 at high pressure is important due to a number of fundamental condensed matter and planetary science [3]. FUKAI and MIZUTANI [4] observed phase transformation experimentally in MoH system, by gradually increasing pressure of up to 5 GPa and temperature up to 500 ��C using in situ resistometry technique. Later on, ANTONOV et al [5] investigated the phase transformation in MoH up to 1000 ��C using the same technique.
Recently, the structural stability of MgH2 was investigated using the DFT technique as implemented in VASP code [6]. No attempts have been made to predict the structural phase transition in MoH,TcH and RuH under high compression. Also the mechanical stability of these hydrides has not been reported yet. The technological applications of these materials and success of ab-initio methods motivated us to analyze the electronic and structural changes in MoH, TcH and RuH. Apart from the structural phase transition, the present work also includes the discussion on the computation of ground state and mechanical properties of MoH, TcH and RuH.
2 Computational details
The present computation of structural stability and phase transition in MoH, TcH and RuH have been carried out using the DFT-based VASP code [7-9], which is appropriate for electronic structure calculations and ab initio molecular dynamics simulations of molecules and solids. Within the framework of DFT [10-12], the first-principles pseudopotential approach has been used to analyze the stability of the ZB (B4) (space group: 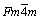 ), WC (Bh) (space group: P6-m2), NaCl (B1) (space group: Fm3m), NiAs (B8) (space group: P63/mmc) and CsCl (B2) (space group: Pm3m) type phases of TcH, MoH and RuH. The unit cells for all five different phases are shown in Fig. 1. The effects of exchange�Ccorrelation interactions are handled by the generalized gradient approximation (GGA). In the present computation, the hydrides are assumed to be without any defect and the stoichiometric composition for all the hydrides is taken as 1:1 ratio of metal atom and hydrogen atom. The electronic wave functions are expanded in a plane wave basis set with an energy cut-off of 450, 500 and 600 eV for MoH, TcH and RuH respectively. The total energy is calculated by integration over a monkhorst-pack mesh [13] of k-point in the Brillouin zone by the linear tetrahedron method including Blochl corrections [8] on the relaxed structures with a smearing width of 0.1 eV. After convergence test, 12��12��12 k-point mesh is chosen to make sure the total energy difference less than 1 meV per primitive cell. The electronic configurations of Mo, Tc, Ru and H atoms are [Kr] 4d55s1 (Z=42), [Kr] 4d55s2 (Z=43), [Kr] 4d75s1 (Z=44), and 1s1 (Z=1) respectively. The valence electronic configurations chosen in our calculation are 4d55s1 for Mo, 4d55s2 for Tc, 4d75s1 for Ru and 1s1 for H atoms.
), WC (Bh) (space group: P6-m2), NaCl (B1) (space group: Fm3m), NiAs (B8) (space group: P63/mmc) and CsCl (B2) (space group: Pm3m) type phases of TcH, MoH and RuH. The unit cells for all five different phases are shown in Fig. 1. The effects of exchange�Ccorrelation interactions are handled by the generalized gradient approximation (GGA). In the present computation, the hydrides are assumed to be without any defect and the stoichiometric composition for all the hydrides is taken as 1:1 ratio of metal atom and hydrogen atom. The electronic wave functions are expanded in a plane wave basis set with an energy cut-off of 450, 500 and 600 eV for MoH, TcH and RuH respectively. The total energy is calculated by integration over a monkhorst-pack mesh [13] of k-point in the Brillouin zone by the linear tetrahedron method including Blochl corrections [8] on the relaxed structures with a smearing width of 0.1 eV. After convergence test, 12��12��12 k-point mesh is chosen to make sure the total energy difference less than 1 meV per primitive cell. The electronic configurations of Mo, Tc, Ru and H atoms are [Kr] 4d55s1 (Z=42), [Kr] 4d55s2 (Z=43), [Kr] 4d75s1 (Z=44), and 1s1 (Z=1) respectively. The valence electronic configurations chosen in our calculation are 4d55s1 for Mo, 4d55s2 for Tc, 4d75s1 for Ru and 1s1 for H atoms.
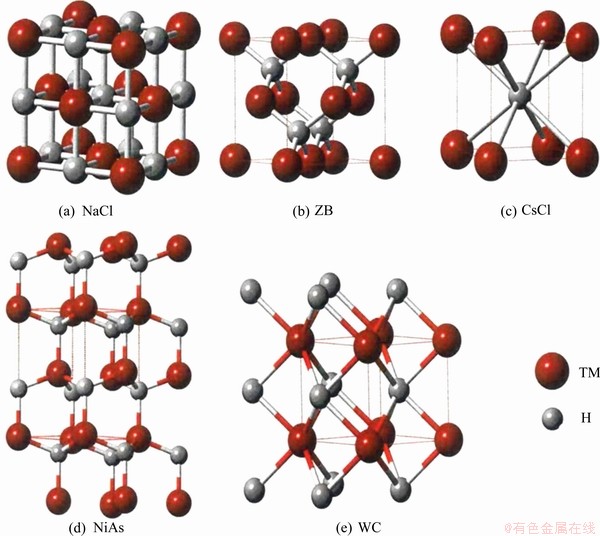
Fig. 1 Unit cell for five different phases of TMH (TM=Mo,Tc,Ru)
3 Results and discussion
3.1 Structural stability and ground state properties
The stability of TMH (TM=Mo, Tc, Ru) is analyzed by calculating the total energy using VASP code based on density functional theory. The computed total energy for all the considered phase of the TMH is listed in Table 1. From Table 1, it is found that both TcH and RuH are energetically stable in the rocksalt (NaCl) structure whereas MoH is energetically stable in the zincblende (ZB) structure. The stability of a solid is determined with the help of its formation energy. The formation energy of a specific compound is defined as the difference between the total energy of the compound and of its constitutive elements. The composition reaction of TMH is as follows:
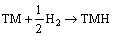 (1)
(1)
This yields the following expression for the formation energy
 (2)
(2)
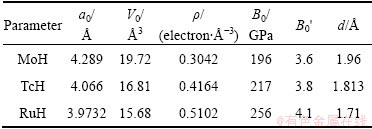
In order to calculate the formation energy, Ef, the total energy of TMH, TM and H2 dimer should be calculated. The formation energies for the transition metals hydrides in five different phases (TMHs) are shown in Fig. 2. From Fig. 2, it is observed that both TcH and RuH are more stable in the NaCl structure whereas MoH is more stable in the ZB structure. The calculated ground state properties like lattice constant a0 (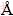 ), cell volume V0(3), valence electron density �� (electron/ 3), bond length (d) of TM��H (), bulk modulus B0 (GPa) and its derivative B��0 for the most stable phase of the TMH are listed in Table 2. Valence electron density (VED) is defined as the total number of valence electrons divided by volume per unit cell and is an important factor for analyzing the super hard materials.
), cell volume V0(3), valence electron density �� (electron/ 3), bond length (d) of TM��H (), bulk modulus B0 (GPa) and its derivative B��0 for the most stable phase of the TMH are listed in Table 2. Valence electron density (VED) is defined as the total number of valence electrons divided by volume per unit cell and is an important factor for analyzing the super hard materials.
Table 1 Total energy of MoH, TcH and RuH
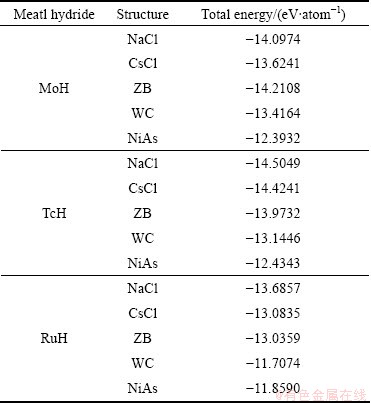
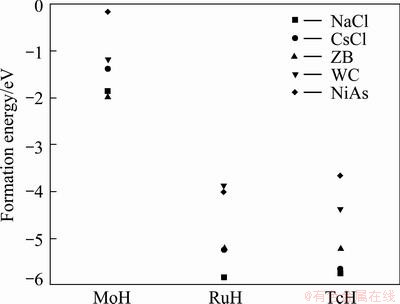
Fig. 2 Formation energy of TMH
Table 2 Calculated ground state properties of MoH, TcH and RuH for five different structures
3.2 Electronic structure
In order to understand the electronic structure of MoH, TcH and RuH, the electronic band structure and density of states (DOS) have been calculated. Figure 3(a) shows the combined band structure and density of states of MoH (ZB), TcH (NaCl) and RuH (NaCl) in their most structure at ambient pressure. The partial density of states (PDOS) for MoH, TcH and RuH is shown in Fig. 3(b). From Fig. 3(a), it is observed that all these hydrides exhibit metallic character and their valence bands split into two parts. The lower part of the valence band is dominated by the ns state electrons of the TM (Mo,Tc,Ru) atoms and slightly influenced by 1s state of the hydrogen atom, while the upper one is a result of strong hybridization from the d state electrons of TM (Mo,Tc,Ru) atoms and the 1s state of H atom. In the density of states of MoH, TcH and RuH (Fig. 3), there is a deep valley called pseudo gap near the Fermi level, which results from the strong hybridization between the transition metal d states and 1s state of H atom. The presence of pseudogap indicates significant covalent bonding between TM and H atoms in the cubic structure.
To further explore the bonding nature, the charge density distribution of MoH, TcH and RuH in the cubic structure is shown in Fig. 4. It is observed that the charge density around H atoms exhibits a strong directional distribution of TM atoms, indicating that the bonding between TM (Mo, Tc, Ru) and H atoms is covalent in nature. The charge density distribution between TM and H atoms in the cubic structure is much denser than those in other structures. Thus the results demonstrate that the bonding in these hydrides is a mixture of metallic, covalent and ionic in attribution.
3.3 Structural phase transition under pressure
At normal pressure MoH is highly stable in zinc blende structure whereas TcH and RuH are stable in the rocksalt structure. The total energy calculations are performed for five different phases of MoH, TcH and RuH, corresponding to the reduced volume of V/Vo=1.0-0.5 and the results are shown in Fig. 5. It is observed that at high pressure, the hexagonal structure becomes more stable in energy than the cubic structure for MoH and TcH. But for RuH, ZB structure is the meta stable phase and hexagonal NiAs is the stable phase at high pressure. Thus it can be concluded that at high pressure all the three hydrides are more stable in the hexagonal phase.
The enthalpy is defined as
H(P)=Etotal(P)+pV(P) (3)
To analyze the structural phase transition from ZB to WC in MoH, from NaCl to NiAs in TcH and from NaCl to ZB to NiAs in RuH, enthalpy values are plotted against pressure and are shown in Fig. 6. The transition pressure estimated from Fig. 6 is tabulated in Table 3. From Table 3 it is concluded that all the three hydrides are stable in the hexagonal phase at high pressure. So far, no study on the phase transition has been performed as a function of high pressure for MoH, TcH and RuH.
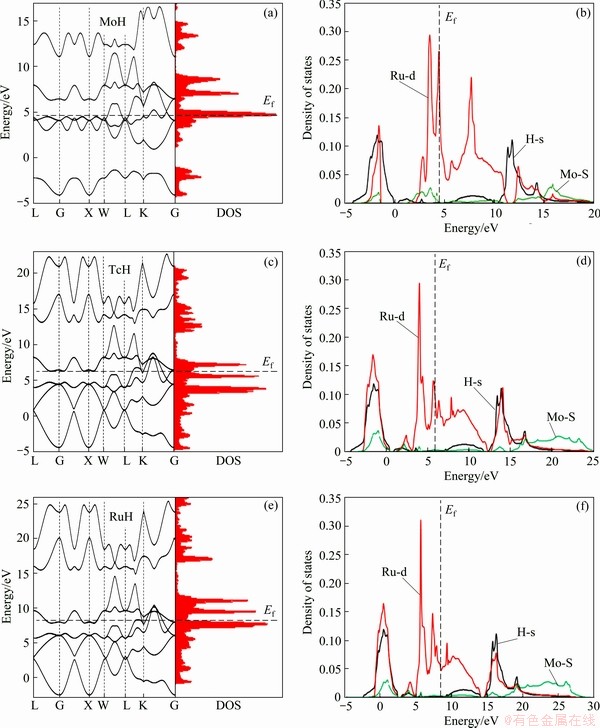
Fig. 3 Electronic band structure (a, c, e) and density and partial density (b, d, f) of states for TMH
3.4 Elastic properties
The elastic constants of solids provide a link between mechanical and dynamical behaviors of crystals, and give important information concerning the nature of forces operating in solids. In particular, they provide information on stability and stiffness of materials [14]. Thus, it is essential to investigate the elastic constants to understand the mechanical properties of MoH, TcH and RuH. Consider a symmetric 3��3 nonrotating strain tensor �� which has matrix elements ��ij (i, j=1, 2 and 3) defined by
�� which has matrix elements ��ij (i, j=1, 2 and 3) defined by
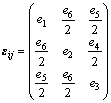 (4)
(4)
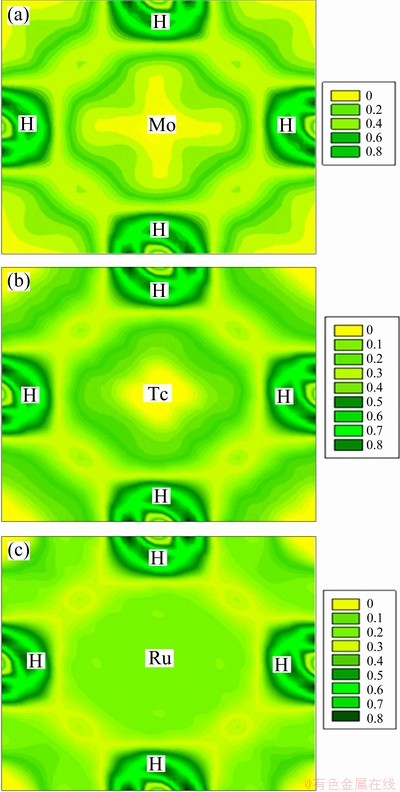
Fig. 4 Charge density distribution of TMHs
Such a strain transforms the three lattice vectors defining the unstrained Bravais lattice {aK, K=1, 2 and 3) to the strained vectors {a��K, K=1, 2 and 3} as given by Eq. (5).
 (5)
(5)
where I is defined by its elements, and Iij =1 for i=j and 0 for i��j. Each lattice vector ak or a��k is a 3��1 matrix. The change in total energy due to the above strain is
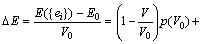
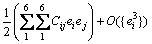 (6)
(6)
where V0 is the volume of the unstrained lattice, E0 is the total minimum energy in this unstrained volume of the crystal, p(V0) is the pressure of the unstrained lattice, and V is the new volume of the lattice due to strain in Eq. (4) where Cij=Cji due to crystal symmetry. This reduces the elastic constants from 36 to 21. Further crystal symmetry reduces the number to 5 (C11, C12, C44, C13, C33) for hexagonal crystals and 3(C11, C12, C44) for cubic crystals. A proper choice of the set of strains (ei, i=1, 2, ��, 6) in Eq. (4) leads to a parabolic relationship between ��E/V0 (��E �� E-E0) and the chosen strain. Such choices for the set ei and the corresponding form for ��E are listed in Table 4 for cubic [15] and Table 5 for hexagonal [16] lattices. For each lattice structure of TMH (TM=Mo, Tc, Ru) studied, the lattice was strained by 0, ��1%, and ��2% to obtain the total minimum energies E(V) at these strains. These energies and strains were fitted with the corresponding parabolic equations of ��E/V0 as listed in Table 4 and 5 to yield the required second-order elastic constants. While computing these energies all atoms are allowed to relax with the cell shape and volume fixed by the choice of strains ei.
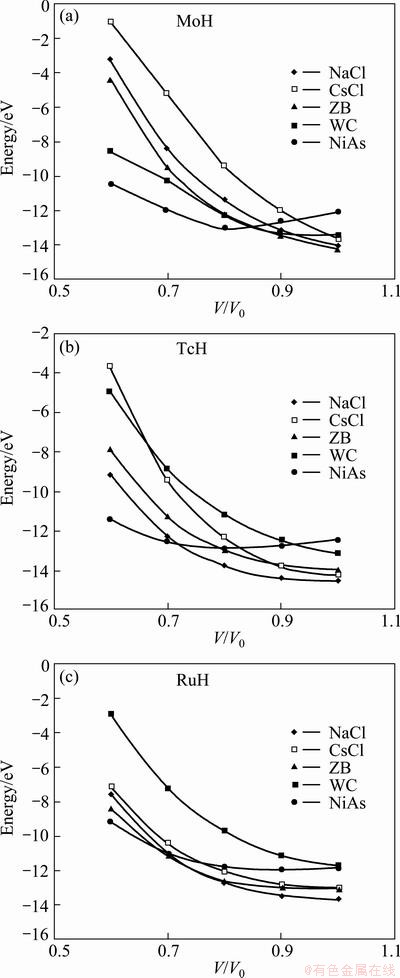
Fig. 5 Total energies as a function of reduced volume
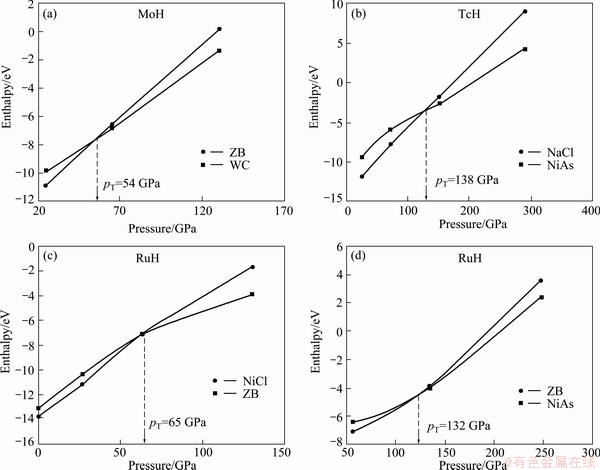
Fig. 6 Enthalpy as function of pressure
Table 3 Estimated transition pressure pT
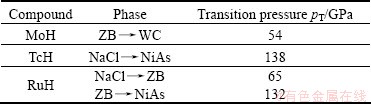
Table 4 Strain combinations in strain tensor by Eq. (4) for calculating elastic constants of cubic structures (rock salt, zinc blende and CsCl)
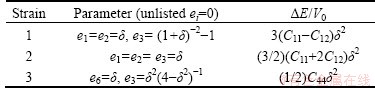
Table 5 Strain combinations in the strain tensor Eq. (4) for calculating elastic constants of hexagonal structures (wurtzite and NiAs)
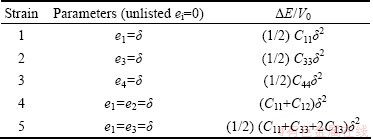
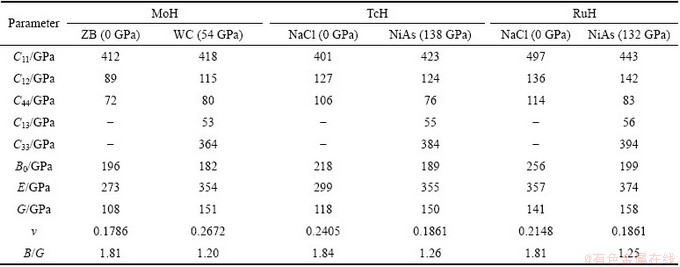
In this work, the calculated single crystal elastic constants Cij, bulk modulus B0, elastic modulus E, shear modulus G, Poisson ratio ��, B/G ratio of TMH (TM= Mo,Tc,Ru) at ambient pressure for cubic structure and high pressure for hexagonal structure are shown in Table 6.
The bulk modulus B0 and shear modulus G for cubic and hexagonal crystal are calculated using the Voigt Reuss-Hill (VRH) approximation [17-19].
The Voigt average for the bulk modulus of the cubic and hexagonal systems respectively is given by:
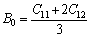 (7)
(7)
 (8)
(8)
The Voigt average for the shear modulus of the cubic and hexagonal systems respectively is given by:
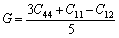 (9)
(9)
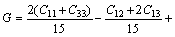
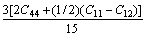 (10)
(10)
Table 6 Calculated elastic constants C11, C12, C44, C13, C33, elastic modulus E, shear modulus G, B/G ratio and Poisson ratio ��
The strain energy 1/2Cijeiej of a given crystal in Eq. (6) must always be positive for all possible values of the set ei, otherwise the crystal would be mechanically unstable. For a stable cubic structure, the three independent elastic constants Cij (C11, C12, C44) should satisfy the well known Born-Huang criteria for the stability of cubic crystals [20].
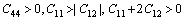 (11)
(11)
while for a hexagonal crystal, the five independent elastic constants Cij (C11, C12, C33, C13, C44) should satisfy the well known Born-Huang criteria for stability [20].
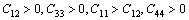 (12)
(12)
 (13)
(13)
Clearly, the calculated elastic constants for cubic and hexagonal TMHs (TM= Mo, Tc, Ru) satisfy Born-Huang criteria, suggesting that they are mechanically stable.
Elastic modulus (E) is calculated in terms of the computed data using the following relation:
 (14)
(14)
B0 and G can measure the resistance of a material to volume and shape change respectively. As listed in Table 6, the results indicate that all the three transition metal hydrides have seemed to more incline to resist with volume change than shape change. Elastic modulus is often used to provide a measure of stiffness of a solid, i.e., the larger the value of elastic modulus, the stiffer the material. The B0, G and elastic modulus of molybdenum hydride are comparable with those of CrH2 [21]. Among these hydrides cubic RuH is stiffer than MoH or TcH.
Poisson ratio is associated with the volume change during uniaxial deformation, which is expressed by Eq. (15) for cubic and Eq. (16) for hexagonal crystals:
 (15)
(15)
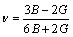 (16)
(16)
During elastic deformation no volume change occurs. If v=0.5, it indicates that the material is incompressible. The low v value means that a large volume change is associated with its deformation. In addition, Poisson ratio provides more information about the characteristics of the bonding forces than any of the other elastic constants. Among the three hydrides, Poisson ratio of cubic MoH is lower than TcH and RuH, indicating that Mo�CH bonding is more directional. The ratio of bulk modulus to shear modulus is used to estimate the brittle or ductile behaviour of materials. A high B/G value is associated with ductility, while a low B/G value corresponds to brittle nature. The critical value which separates ductile and brittle materials is about 1.75. From Table 6, it is found that cubic MoH,TcH and RuH are brittle in nature.
4 Conclusions
1) Cubic structure is the most stable structure at ambient pressure.
2) All the calculated elastic constants obey the Born-Huang criteria, suggesting that they are mechanically stable.
3) It is observed that the bonding in cubic MoH, TcH and RuH structure is a mixture of metallic, covalent, and ionic characters.
4) A pressure induced structural phase transition from cubic to hexagonal phase is also predicted in MoH, TcH and RuH. This work can stimulate research on MoH, TcH and RuH.
References
[1] ZUTTEL A. Materials for hydrogen storage [J]. Mater today, 2003, 6(9): 24-33.
[2] TAPPING R L. GENDRON T S. Atomic energy of Canada limited, Report No.RC-101/COG-88-136 [R].1988-9.
[3] MAO H K. HEMLEY R J. X-ray-induced dissociation of H2O and formation of an O2-H2alloy at high pressure [J]. American Scientist, 1992, 80: 234-247.
[4] FUKAI Y, MIZUTANI M. The phase diagram of Mo-H alloys under high hydrogen pressures [J]. Mater Trans, 2003, 44(7): 1359-1362.
[5] ANTONOV V E, BELASH I T, PONYATOVSKII E G. T�CP phase diagrams and isotope effects in the Mo�CH/D systems [J]. Dokl Akad Nauk SSSR, 1979, 248(3): 635-637.
[6] KANAGAPRABHA S. ASVINI MEENAATCI A T, RAJESWARA PALANICHAMY R, IYAKUTTI K. First principles study of pressure induced structural phase transition in hydrogen storage material��MgH2 [J]. Physica B, 2012, 407(1): 54-59.
[7] PERDEW P, BURKE S. Generalized gradient approximation for the exchange-correlation hole of a many-electron system [J]. Physical Review B, 2004, 54(23): 16533.
[8]  P E. Projector augmented-wave method [J]. Physical Review B, 1994, 50(24): 17953.
P E. Projector augmented-wave method [J]. Physical Review B, 1994, 50(24): 17953.
[9] KRESSE G, JOUBERT. From ultrasoft pseudopotentials to the projector augmented-wave method [J]. Physical Review B, 1999, 59(3): 1758.
[10] KRESSE G, HAFNER J. Ab initiomolecular dynamics for liquid metals [J]. Physical Review B, 1993, 47(1): 558.
[11] KRESSE G, FURTHMULLER. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set [J]. Computational Material Science, 1996, 6: 15-50.
[12] PERDEW J P, BURKE S. Errata: Generalized gradient approximation made simple [J]. Physical Review Lett, 1997, 78(7): 1396.
[13] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Physical Review B, 1976, 13(12): 5188.
[14] NYE J F. Physical properties of crystals. Their representation by tensors and matrices Chap VIII [M]. Oxford Press, 1957.
[15] KALAY M, KART H H, CAGIN T. Elastic properties and pressure induced transitions of ZnO polymorphs from first-principle calculations [J]. Journal of Alloys and Compounds, 2009, 484(1-2): 431-438.
[16] GERD STEINLE-NEUMANN, LARS STIXRUDE, RONALD E.COHEN. Erratum: First-principles elastic constants for the hcp transition metals Fe, Co, and Re at high pressure [J]. Physical Review B, 2004, 69(21): 219903.
[17] VOIGT W. A determination of the elastic constants for beta-quartz Lehrbuch de Kristallphysik (Terubner, Leipzig, 1928).
[18] REUSS A, ANGEW Z. Berechnung der Flie��grenze von Mischkristallen auf Grund 32der Plastizit��atsbedingung f��ur Einkristalle [J]. Zeitschrift f��r Angewandte Mathematik und Mechanik,1929, 9(1): 49-58.
[19] HILL R. The elastic behaviour of a crystalline aggregate [J]. Proceedings of the Physical Society, London A, 1952, 65: 349.
[20] BORN M, HUANG K. Dynamical theory of crystal lattices [M]. Oxford and New York: Clarendon Press, 1988.
[21] CHIHI T, FATMI M, BOUHEMADOU A, Structural, mechanical and electronic properties of transition metal hydrides MH2 (M=Ti, Zr, Hf, Sc, Y, La, V and Cr) [J]. Solid State Sciences, 2012, 14: 583-586.
���ɽ����⻯��TMH(TM=Mo��TC��RU)������֯���������Ժ���ѧ���ܵĵ�һ��ԭ���о�
G. SUDHA PRIYANGA1, A.T.ASVINI MEENAATCI1, R. RAJESWARA PALANICHAMY1, K.IYAKUTTI2
1. N.M.S.S. Vellaichamy Nadar College, Madurai, Tamil nadu-625019, India;
2. Department of Physics and Nanotechnology, SRM University, Chennai, Tamil nadu-603203, India
ժ Ҫ��ͨ�����ڹ����ݶȽ����ܶȷ������۵ĵ�һ��ԭ���о����ɽ����⻯��TMH(TM =Mo��TC��RU)������֯���������Ժ���ѧ���ܡ��ڱ��о���5������ṹ�У������ṹ���������ṹ���ȶ���Ԥ���˲�ͬ���ɽ����⻯���ڸ�ѹ�µ���ṹת�䣬�磺MoH�д�ZB�ṹ��WC�ṹ��ת�䡢��TcH�д�NaCl�ṹ��NiAs�ṹ��ת���Լ���RuH�д�NaCl�ṹ��ZB�ṹ�ٵ�NiAs�ṹ��ת�䡣���Գ����ļ�������������⻯�����ѧ�����ڻ���ѹ�������ȶ��ġ�
�ؼ��ʣ���һԭ�����ṹ��䣻�������ԣ���������
(Edited by Chao WANG)
Corresponding author: R. RAJESWARA PALANICHAMY; Tel: +452-2459187; Fax: +452-2458358; E-mail: rajeswarapalanichamy@gmail.com
DOI: 10.1016/S1003-6326(13)62787-9
Abstract: The structural, electronic and mechanical properties of transition metal hydrides (TMH, TM=Mo, Tc, Ru) are investigated by means of first principles calculation based on density functional theory with generalized gradient approximation. Among the five crystallographic structures that have been investigated, the cubic phase is found to be more stable than the hexagonal ones. A structural phase transition from ZB to WC in MoH, NaCl to NiAs in TcH and NaCl to ZB to NiAs in RuH is also predicted under high pressure. The calculated elastic constants indicate that all the three hydrides are mechanically stable at ambient pressure.
