
���±�ţ�1004-0609(2009)08-1505-06
Au/AC�������Ʊ����������
�� ��1��������1����Ż�2�������3��ʷ ��3
(1. �廪��ѧ ������ѧ�빤��ϵ������ 100084��
2. �����Ƽ���ѧ ��ľ�뻷������ѧԺ������ 100083��
3. �����Ƽ���ѧ ұ������̬����ѧԺ������ 100083)
ժ Ҫ��
ժ Ҫ�����ý��շ��Ʊ����Ի���̿���ؽ����(Au/AC)����ͨ��N2����?�Ѹ������������(XPS)�Ըô������б���������Au/AC�����������¶Ե�Ũ�ȳ������ױ��Լ����߹���ʱ�Ĵ��ֽ����ܡ����������Au/AC�����Գ������������Ĵ��ֽ���ԣ������¡�����76 000 h?1�����ʪ��(45��5)%�����£�Au/AC�Գ�ʼŨ��Ϊ(55��5) mg/m3 �ij�����2 300 minʱ��ȥ����Ϊ91.3%��Au/AC�Լױ���ȥ��δ���ֳ����ԵĴ��ֽ����ܣ���������������Ϊ�����ڳ����ͼױ�����������£�Au/AC�Գ����Ĵ��ֽ�����������ͣ��������Ӧ��Au/AC�ı���ʯī̼�����������ͣ�˵�������ܹ�ͨ��Au/AC���������̿�ķ�Ӧ���Խ��⣬���ڷ�Ӧǰ�������Au�ܹ������ȶ��ĵ��ʼ�̬��
�ؼ��ʣ�
��ͼ����ţ�O 643 ���ױ�ʶ�룺 A
Preparation and catalytic activity of Au/AC catalyst
ZHANG Bo1, ZHANG Peng-yi1, XU Jiu-hua2, LI Hong-xu3, SHI Rui3
(1. Department of Environmental Science and Engineering, Tsinghua University, Beijing 100084,China;
2. School of Civil and Environmental Engineering, University of Science and Technology Beijing,
Beijing 100083, China;
3. School of Metallurgical and Ecological Engineering, University of Science and Technology Beijing,
Beijing 100083,China)
Abstract: The catalysts of Au supported on modified activated carbon were prepared by impregnation method and characterized by N2 adsorption-desorption and XPS. The catalytic activity of Au/AC for decomposition of ozone and toluene was evaluated. The results show that, under the condition of (55��5) mg/m3 ozone in air, space velocity 76 000 h?1, relative humidity (45��5)% and ambient temperature, the ozone removal ratio of the catalyst can be maintained at 91.3% within 2 300 min. And the activity of Au/AC catalyst is reduced when the toluene exists in the air. The graphitic carbon content of Au/AC decreases dramatically, which indicates that Au/AC can promote the reaction betweens the ozone and carbon. For the reason of competitive adsorption when ozone and toluene coexist, the activity of Au/AC declines obviously. However, Au maintains its stable state of metallic gold before and after the reaction.
Key words: gold catalyst; catalysis; ozone; impregnation
��20����80������ڣ�HARUTA��[1]���ָ����ڹ��ɽ����������ϵ���������Ե���CO�������нϸߵĴ����Ժ����������˶�����������о��ȳ������������ؽ�����ڵ���CO����[2?5]��ϩ�������л��ﻷ����[6?7]�������ȼ��[8?9]�Լ�����ˮ����Ӧ[10]�ȷ���Ĵ������ܵ��㷺��ע��HAO��[11]������Ȫ��[12]�ֱ��о��˽����������ؽ�����Գ�����CO��ȥ��Ч����ROSSI��[13]��CARRETTIN��[13?14]�ֱ��Ʊ��˸�����Ϊ0.8%��X40S��Ҭ�ǻ���̿�ؽ����������Ϊ1%��XC72R��ʯ���Ի���̿�ؽ�����������������Ƕ��Ǻ��Ҵ�Һ�������Ĵ����ԡ�������̿�ؽ�����������¶Ե�Ũ�ȳ����Ĵ��ֽ���ԡ��Լ��ױ�������Ե�Ӱ�컹�ʼ����������������Խ��;����ڵij���Ũ��ΪĿ�ģ����������ڿ����ĸ����Զ�����ӷ����л���Ⱦ��ױ����о��Ը��Ի���̿Ϊ���壬���ü����еĽ��շ��Ʊ�Au/AC�������������ִ����Գ����Ĵ��ֽ����ܡ��Լ��ױ��Ľ����������Ե�Ӱ�졣
1 ʵ��
1.1 �������Ʊ�
��ɽ���»�������������DX09��ú�ʻ���̿(��ΪAC)Ϊ���壬������и��Դ���������̿����10%��ϡ������60 ��ˮԡ����2 h������2.5 mol/L NaBH4�����½���4 h��ϴ�����Բ���ɱ���(��ΪM-AC)�����Ƚ���(HAuCl4��4H2O)Ϊ��ǰ������Һ���Ҷ���(En)Ϊ�������䰴������1?1���ƽ���Ϊ 5 g/L��ˮ��Һ��Ϊ����Һ����5 g���Ժ�Ļ���̿���յ�10 mL����Һ�У���������2 minʹ���ɢ���ȣ����˳�����̿�ڳ��������ɣ�����200 ������H2��ԭ40 min�����Ƶû���̿�ؽ����(��ΪAu/AC)��
1.2 �����ı���
���õ�����?�Ѹ���(QuadraSorb SI������Ϊ�����ʣ����ѹ��p/p0Ϊ10?6~1)�ⶨ�����ıȱ�����Ϳ�϶�ṹ�����ù����������(PHI-5300 ESCA��Al/MgΪ˫�����У�����C1s(����Ϊ284.8 eV))���кɵ�У�����ⶨ���������Ԫ����ɼ���̬��
1.3 ���������ܲ���
�������Բ����ڳ��³�ѹ�£�������䴲����������ʽ�������ۡ����ھ�Ϊ2 cm�IJ���ֹ���װ��1.6 g������������Ϊ0.8 cm����������Ϊ4 L/min����Ӧ�Ŀ���Ϊ96 000 h?1���Ӵ�ʱ��Ϊ0.04 s����������Ũ��Ϊ(55��5)mg/m3�����������ʪ��Ϊ(45��5)%������Ũ�ȼ����ʪ�ȷֱ��������ȵ繫˾��Model 49C�ͳ��������ǡ�Mannix Lan 880D����ʪ�ȼƲⶨ�������Ļ�������Գ�����ȥ�����Լ�����ʱ������ʾ���ױ���ʼŨ��Ϊ3.8 mg/m3��������ɫ����HP5890H/PH�����ⶨ��ͨ����ͨ��ֱ�ӽ�����������Ϊ0.5 mL��ɫ���������£������ΪOV?101������N2��ѹ��Ϊ80 kPa���������¶�150 �棬����90 �棬������¶�200 �档
2 ���������
2.1 ������������
2.1.1 Au/AC�Գ������ֽ����
ͼ1��ʾΪAC��M-AC��Au/AC��3����Ʒ�Գ����ķֽ����ܡ��Ƚ����߶Գ�����ȥ�������߿�֪��Au/AC�����Գ����Ĵ��ֽ�����������������AC��M-AC�Ĵ��ֽ����ܣ���1 400 minʱ��Au/AC��ȥ���ʱ�����95%���ϣ�����ʱAC��M-AC�Գ�����ȥ�����ѷֱ��½���72%��78%���ɴ˿ɼ������ػ�����ֽ��Ƴɵ�Au/AC������δ���ص�AC��M-AC��ȣ�Au/AC�Գ�����ȥ�����ܴ������ߣ�˵�����صĻ�����ֽ���д��ֽ�����Ļ��ԡ��Ա�AC��M-AC�Գ�����ȥ�����߿�֪��M-AC�Գ����ķֽ�����������AC�ģ�˵����AC���и��Դ�����������߶Գ�����ȥ��������

ͼ1 AC��M-AC��Au/AC�Գ����ķֽ�����
Fig.1 Decomposition ability of AC, M-AC and Au/AC catalyst for ozone
2.1.2 Au/AC�Լױ���ȥ������
ʵ���п���������������ʱAu/AC�Լױ���ȥ��������ͼ2��ʾΪAC��M-AC��Au/AC�Լױ���ȥ����������ͼ2���Կ�����3�����ߵ��½��������ƣ������Բ�ͬ��ͼ1��3������ȥ�����������ֳ�����ʱ�����ƻ����½������ơ��ڱ��������У���Ӧǰ��3����Ʒ�Լױ������ֽϸߵ�ȥ��Ч�ʣ��ҳ���ƽ���½������ƣ�������ʱ����ӳ�����ijһʱ��ȥ����ͻȻ�����½���20%���ҡ�����ȥ�����������ֳ������������Ϲ�?�������������ߵ����ԣ���ˣ��ڴ˷�Ӧ�У���������Ӧ���ڴ��ֽ����á�AC��ǰ1 051 min�ڶԼױ���ȥ��������ƽ���½��ڣ���ʱAC�Լױ���ȥ����������M-AC��Au/AC�ģ���ȥ��Ч����100%ƽ���½���96%����1 051 min�ױ�ȥ����Ѹ���½�����2 054 minʱ���½���15%����M-AC��Au/ACȥ���ױ���ƽ���½��ھ������ӳ����ֱ���1 364 min��1 890 min�����½���AC��M-AC��Au/AC�ڽ�1 850 minʱ�Լױ���ȥ���ʷֱ��½���38.4%��25.1%��83.3%�������ڴ�ʱ����3����Ʒ�Լױ���ȥ�����ֱ�Ϊ17.16��17.31��26.32 mg����˵��Au/AC�Լױ���ȥ����Ȼ����������Ϊ��������ȥ���ױ�����ǿ�ڸ���ǰ��AC��M-AC�����صĽ��������һ�����ֽ�ױ���������

ͼ2 AC��M-AC��Au/AC�Լױ���ȥ������
Fig.2 Decomposition ability of AC, M-AC and Au/AC catalysts for toluene
2.1.3 ������ױ�����ʱAu/AC�Ĵ����Բ���
ͼ3��ʾ�ֱ�Ϊ�����ͼױ�����ʱAC��M-AC��Au/AC��������Ⱦ���ȥ�������ߡ���ͼ3(a)��֪�����мױ�����ʱ��3����Ʒ�Գ�����ȥ�������߱��ֳ�һ�µ��½����ƣ���ͼ1��ʾ�Ľ����ȣ�3����Ʒ�Գ�����ȥ�������������½�������Au/ACΪ���� Au/AC��δ���ֳ��Գ����Ĵ��ֽ�����Զ����AC��M-AC�����ƣ�˵���ױ��Ľ��벻����Au/AC�Գ����Ĵ��ֽ⣬������Au/AC�Ĵ����ԡ��Ա�ͼ3(b)�е�3���ױ�ȥ�������߿��Կ��������ߵ��½��������Բ�ͬ��AC��M-AC�ױ�ȥ�������ߵ��½�������ͼ2��3���������ƣ��Ա��ֳ���?�������������ߵ����ԣ�˵���ڳ����ͼױ�����������£�AC��M-AC�Լױ���ȥ��������������Ϊ������Au/AC�ļױ�ȥ���������½�����ȴ�����仯���淴Ӧʱ������Ӽױ���ȥ����һֱ����ƽ���½����ƣ���˵����������Ⱦ���ʹ��������£�Au/AC�Լױ���ȥ�����ɵ�һ�ױ�����ʱ����������Ϊ����ת��Ϊ�Դ��ֽ�����Ϊ���������ϲ��Խ��������Ϊ�����мױ�����ʱ�����ڼױ��ͳ�����Au/AC�Ľ����λ���ھ����������ã��������������ֽ��������Ч����λ�����������٣�ʹ��Գ����ķֽ����������½���ͬʱAu/AC��Ԫ����ɺͿ�϶�ṹ���ڳ����Ĵ��ֽⷴӦ�������仯��ʹ��Լױ�����������Ҳ��͡��ڳ����ͼױ����������£�Au/AC�ܹ��ٽ�һ���ּױ������������Ӧ�����¼ױ�����Ҫȥ�����Ʒ����ı䡣
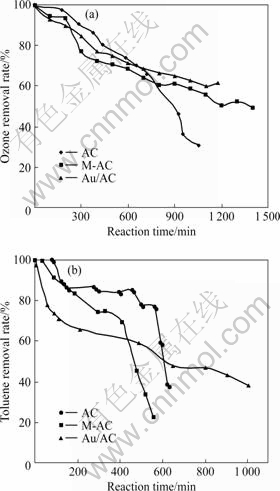
ͼ3 ������ױ�����������AC��M-AC��Au/AC�Զ��ߵ�ȥ������
Fig.3 Decomposition ability of AC, M-AC and Au/AC catatysts for ozone (a) and toluence (b) at coexisting ozone and toluene
2.2 ��������
2.2.1 N2����?�Ѹ�����
��AC��M-AC��Au/AC��3����Ʒ������N2����?�Ѹ�����������N2����?�Ѹ���������õ�����Ʒ�ıȱ���������ݺͿ���������ڱ�1���ɱ�1�ɿ�������ƷAC��M-AC��Au/AC��BET�ȱ�������ܿ��ݾ������½�����ƽ���������ߡ���������AC�ڸ��Թ����У����϶�ṹ��ƻ�����ɲ��ֿ�϶�����ṹ̮������ʹ�ȱ�������ܿ����½������ؽ��Ժ��Au/AC�ȱ�������ܿ��ݽ�һ���½������ǵ����ԭ�Ӱ뾶Ϊ0.144 nm[15]������Ϊ2 nm�������ڻ���̿����Ŀ�϶�����ռ�нϴ��������ˣ������ƶϣ���������������뵽��������е��²������������Աȱ�������ܿ��ݵĹ���������Ե�����������ʱ����Ȼ���±ȱ�������ܿ����½���ƽ�������ӡ�
��1 �����������Ʒ��N2����?�Ѹ������
Table 1 Results from N2 adsorption-desorption measurement of catalysts AC, M-AC and Au/AC

2.2.2 XPS����
�ֱ��δ�μӷ�Ӧ�Ĵ���Au/AC���������Ӧ���Au/AC(���Au/AC/O3)����ױ���Ӧ���Au/AC(���Au/AC/C7H8)�Լ��ڳ������ױ����������·�Ӧ���Au/AC(���Au/AC/(O3+C7H8)) 4����Ʒ����XPS������ͨ������Ʒ���溬����������ٷֺ����仯����Ŀ��죬��һ������Au/AC�Ĵ����û��ơ�C1s�������ɽ���Ϊ5�ֶ����ķ壬�ֱ�Ϊ��ʯī̼���ӡ������ѵ�C��OH���ʻ�������C=O���Ȼ�������COOR�Լ���-��*����Я����[16?17]������Ʒ�Ľ���������2���С��Ƚ�4����Ʒ�к��������ŵ���Ժ������Կ�������δ�μӷ�Ӧ��Au/AC��ȣ�������3�ַ�Ӧ�����Ʒ�У�Au/AC/C7H8�������ŵ���Ժ����仯������С���������漸��û�з�����Ӧ�����ͼ2�жԼױ�ȥ���������ص�ķ�������һ��֤����Au/AC�Լױ���ȥ��������������Ϊ����Au/AC/O3����������Ժ����仯�������ʯī����Ժ���̼��70.1%�½���54.1%��COOR����Ժ���̼��4.3%������13.9%��˵���ڳ������ֽⷴӦ�������淴Ӧʱ�����ӣ�ʯī̼��C��OR��C=O��������������������COOR���ڲ��ױ���һ��������CO��CO2�������ۻ��������京����������ӡ�Au/AC/(O3+C7H8)��Ʒ�и������ŵ���Ժ����仯���Ƚ���ǰ����֮�䣬ԭ���ǵ��мױ�����ʱ�������ͼױ���Au/AC����Ļ���λ���ھ���������һ���ֻ���λ���������˼ױ���ɥʧ�Գ����Ĵ��ֽ���ԣ�ʹ�������ֽⷴӦ����Ч����λ�������٣��Ӷ����������������Ӧ��̼Ԫ�ص���Ҳ��Ӧ���١���ˣ���������Ⱦ�ﹲ��������£���Au/AC/O3��ȣ�Au/AC/(O3+C7H8)�ı���ʯī̼�����������٣��Գ�����ȥ�������½���ͬʱ�������ڴ��ֽ������Ӧ��������������Ԫ����ɺͿ�϶�ṹ�ĸı䣬����Au/AC/(O3+C7H8) �Լױ�����������ҲԶ����Au/AC/C7H8�ġ�
ͼ4��ʾΪ4����Ʒ����Au4f��XPS���ס�Au/AC��Ʒ����Au4f7/2�Ľ����λ��Ϊ84.2 eV����Ӧ��Ԫ�ػ�ѧ̬Ϊ���ʽ�[18]����ͼ4�ɿ���������3�ֲ�ͬ��Ӧ���̺���ƷAu/AC/O3��Au/AC/C7H8 ��Au/AC/(O3+C7H8)����Au 4f�Ľ���ܲ�û�з�������λ�ƣ�˵���������Au�ڷ�Ӧǰ���ܹ������ȶ��ĵ��ʼ�̬����ˣ�������ܲ��Խ���ͱ������������Ϊ��Au/AC�ܹ�ͨ�������������̿�ķ�Ӧ��ʵ�ֶԳ����Ľ��⣬���ڷ�Ӧ������Au�ܹ������ȶ��ĵ��ʼ�̬��
��2 ��ͬ��Ʒ��C1s XPS����Ͻ��
Table 2 Fitting results of C1s XPS spectra of different samples


ͼ4 ��ͬ��Ʒ��Au4f XPS��
Fig.4 Au4f XPS spectra of different samples
3 ����
1) ���Ҷ���Ϊ���屣�������ý��շ��Ʊ��ĸ��Ի���̿�ؽ�����������������£����ֽ�Ũ��Ϊ(55��5)mg/m3�ij�������2 300 minʱ���Գ�����ȥ����Ϊ91.3%��XPS������ʾ��Au/AC�ڴ������ֽⷴӦ��������ʯī̼��ٷֺ������Խ��ͣ�������ֽ��ڷ�Ӧǰ��ʼ�ձ����ȶ��ĵ���״̬��˵��Au/AC�ܹ�ͨ�������������̿�ķ�Ӧ��ʵ�ֶԳ����Ľ��⡣
2) �ڳ�����ױ�����������£����ڼױ��Ľ���ʹAu/AC���ڴ��ֽ��������Ч����λ�������٣�����Au/AC�Գ�����ȥ�������Խ��͡�ͬʱ��������̼�ķ�Ӧ���ƻ���Au/AC�ı���Ԫ����ɺͿ�϶�ṹ�Ӷ������˶Լױ����������ܡ�
[1] HARUTA M, YAMADA N, KOBYSAHI T, LIJIMA S. Gold catalysts prepared by coprecipitation for low-temperature oxidation of hydrogen and of carbon monoxide[J]. J Catal, 1989, 115(2): 301?309.
[2] BULUSHEV D A, YURANOV I, SUVOROVA E I, BUFFAT P A, KIWI-MINSKER L. Highly dispersed gold on activated carbon fibers for low-temperature CO oxidation[J]. J Catal, 2004, 224(1): 8?17.
[3] ����ѧ, ����, ��ѩ÷, ������. Au/TiO2�������Ʊ����������[J]. ��ѧͨ��, 2005(10): 785?788.
QI Shi-xue, ZOU Xu-hua, LIU Xue-mei, AN Li-dun. The preparation and photocatalytic activity of Au/TiO2[J]. Chemistry, 2005(10): 785?788.
[4] PILLAI U R, DEEVI S. Highly active gold-ceria catalyst for the room temperature oxidation of carbon monoxide[J]. Appl Catal A, 2006, 299(2): 266?273.
[5] DAT? M, IMAI H, TSUBOTA S, HARUTA M. In situ measurements under flow condition of the CO oxidation over supported gold nanoparticles[J]. Catal Today, 2007, 122(3/4): 222?225.
[6] CHOWDHURY B, BRAVO-SUAREZ J J, DATE M, TSUBOTA S, HARUTA M. Trimethylamine as a gas-phase promoter: Highly efficient epoxidation of propylene over supported gold catalysts[J]. Angew Chem, 2005, 118(3): 426?429.
[7] DIMITRATOS N, LOPEZ-SANCHEZ J A, LENNON D, PORTA F, PRATI L, VILLA A. Effect of particle size on monometallic and bimetallic (Au, Pd)/C on the liquid phase oxidation of glycerol[J]. Catal Lett, 2006, 108(3/4): 147?153.
[8] WATERS R D, WEIMER J J, SMITH J E. An investigation of the activity of coprecipitated gold catalysts for methane oxidation[J]. Catal Lett, 1995, 30(1/4): 181?188
[9] MIAO S J, DENG Y Q. Au-Pt/Co3O4 catalyst for methane combustion[J]. Appl Catal B, 2001, 31(3): L1?L4.
[10] ������, ֣ ��, ������, κ��þ. �������Ե���ˮú���任Au/��-Fe2O3�����ṹ�����ܵ�Ӱ��[J]. ȼ�ϻ�ѧѧ��, 2003, 31(6): 558?563.
HUA Jin-ming, ZHENG Qi, LIN Xing-yi, WEI Ke-mei. Influence of Au loading on the structure and catalytic performance of Au/��-Fe2O3 catalysts for low temperature water-gas shift reaction[J]. Journal of Fuel Chemistry and Technology, 2003, 31(6): 558?563.
[11] HAO Z P, CHENG D Y, GUO Y, LIANG Y H. Supported gold catalysts used for ozone decomposition and simultaneous elimination of ozone and carbon monoxide at ambient temperature[J]. Appl Catal B, 2001, 33(3): 217?222.
[12] ����Ȫ, ������, �¿���, �� ��, ����ѧ, ����. LaFeO3���κ�O3������Au/Al2O3������CO������Ӧ�����ȶ��Ե�Ӱ��[J]. ��ѧ��, 2008, 29(6): 506?508.
LIN Qing-quan, AN Li-dun, CHEN Jun-yong, QIN Hua, QI Shi-xue, ZOU Xu-hua. Effect of LaFeO3 decoration and ozone treatment on the thermal stability of Au/Al2O3 for CO oxidation [J]. Journal of Catalyst, 2008, 29(6): 506?508.
[13] PORTA F, PRATI L, ROSS MI, SCARI G. New Au(0) sols as precursors for heterogeneous liquid-phase oxidation catalysts[J]. J Catal, 2002, 211(2): 464?469.
[14] CARRETTIN S, MCMORN P, JOHNSTON P, GRIFFIN K, HUTCHINGS G J. Selective oxidation of glycerol to glyceric acid using a gold catalyst in aqueous sodium hydroxide[J]. Chem Commun, 2002, 2(7): 696?697.
[15] DEAN J A. Lange��s handbook of chemistry[M]. 15th Ed. New York: McGraw-Hill Press, 1999: 31.
[16] WU S H, PENDLETON P. Adsorption of anionic surfactant activated carbon: Effect of surface chemistry, ionic strength, and hydrophobicity[J]. J Colloid Interface Sci, 2001, 243(2): 306?315.
[17] SUBRAHMANYAM C, BULUSHEV DA, KIWI-MINSKER L. Dynamic behaviour of activated carbon catalysts during ozone decomposition at room temperature[J]. Appl Catal B, 2005, 61(1/2): 98?106.
[18] MINIC? S, SCIR? S, CRISAFULLI C, GALVAGNO S. Influence of catalyst pretreatments on volatile organic compounds oxidation over gold/iron oxide[J]. Appl Catal B, 2001, 34(4): 277?285.
������Ŀ��������Ȼ��ѧ����������Ŀ(50772058)�������ص�ʵ����ר�����������Ŀ(08Y02ESPCT)
�ո����ڣ�2008-11-10�������ڣ�2009-04-17
ͨѶ���ߣ������壬���ڣ���ʿ���绰��010-62796840-601��E-mail: zpy@tsinghua.edu.cn
(�༭ ������)
