
���±�ţ�1004-0609(2010)04-0784-04
���Ӷ���ѧģ��Pb-Au�Ͻ������ѧ����
�ֹ��������£��� ������
(����������ѧ ���ұ����ҹ���ʵ���ң����� 650093)
ժ Ҫ��
���÷��Ӷ���ѧ����ģ��Pb8Au2��Pb9Au1���ֺϽ���ϵ������ѧ���ʡ����㲻ͬ�¶��ºϽ���ϵ�������ʡ����������ܺ�ʣ�����ܣ�ͬʱ����Ͻ�Ľ���ܣ��Ӻ�ۺ��۽Ƕȷ�����Ԫ�Ͻ�ԭ�Ӽ�����ã��õ��ļ�������ʵ�������Ǻϵúܺá�
�ؼ��ʣ�
Pb-Au�Ͻ���Pb8Au2�Ͻ���Pb9Au1�Ͻ������Ӷ���ѧ������������������������ʣ���������������
��ͼ����ţ�TF13���� ���ױ�־�룺A
Thermodynamic properties of Pb-Au alloy simulated by molecular dynamics
JIA Guo-bin, LIU Yuan-yuan, YANG Bin, LIU Da-chun
(National Engineering Laboratory of Vacuum Metallurgy, Kunming University of Science
and Technology, Kunming 650093, China)
Abstract: The molecular dynamics method was used to simulate the thermodynamic properties of two binary alloys, Pb8Au2 and Pb9Au1. The formation enthalpy, free energy of formation and excess free energy of two alloys were calculated. The atomic interactions were analyzed in macroscopic and microcosmic view. The values obtained here are consistent well with those of the experiments.
Key words: Pb-Au alloy; molecular dynamics (MD); formation enthalpy; free energy of formation; excess free energy; cohesive energy
��Ǧ����ɫұ���������ڳ����ĺϽ�ͭ�������Ǧ�����ദ�������ж�������úϽ𡣹�Ǧ����Ҫ����Ag��Au��Cu��Pb��Ԫ�أ���������������ܹ��ԸúϽ������Ч���룬�����еĹ����Au��Ag������Ч��������������ԭ�������úϽ��и��������ѹ�IJ�ͬ��ͨ�����Ƚ�����ѹ��Ľ����ӺϽ��з����ȥ[1]����ۺ�������ѧ���ݶ����жϺϽ��������ķ���̶��ؼ����ã�HAGER��WILKOMIRSKY[2]ͨ������õ�1 000 K��1 200 KʱPb-Au�Ͻ�Ĺ�ʣ�����ܣ�KLEPPA[3]�õ���873 KʱPb-Au�Ͻ�Ĺ�ʣ�����ܡ������ڸ�����������ѧ����ĸ����ԣ�ĿǰPb-Au�Ͻ������ѧ������Ȼ���٣���������Ŀǰ����������ۼ����Ҫ��������ͨ�����Ӷ���ѧ����ģ��298~1 498 KʱPb-Au�Ͻ������ѧ���ʣ����ڶԺ�����������������������ָ������ģ�������ѡȡPb8Au2��Pb9Au1���ֺϽ���ϵ���������ʵ�GEAM�ƣ�ģ�ⲻͬ�¶���������ϵ�ĺ�ۺ�������ѧ���ʡ�
1 ���Ӷ���ѧ���ܺ���
DAW��BASKES[4]�Լ�FOILS��[5]��20����80����������Ƕ��ԭ�ӷ���(EAM)���÷����������ܶȷ������ۣ���һ�ֽ�����ԭ�Ӳ�εİ뾭������ģ�͡��˺�Ƕ��ԭ��ģ�ͼ����ָĽ���Ƕ��ԭ��ģ�͵õ����ٷ�չ[6-8]��Ƕ��ԭ��ģ��[9-14]�ھ�ȷ�о���ϵ������ѧ�Ͷ���ѧ���ʷ��淢�Ӻܴ�����á�
���Ӷ���ѧģ��Ĺؼ�����ȷ��ѡ������ģ�����������Ӽ�����á���WADLEY��[15]��ZHOU��[16]���������Ƕ��ԭ��ģ��(GEAM��)�����������ɽ����ͺϽ�ķ��Ӷ���ѧģ�⣬�ǽ������������������������ԭ�Ӽ�����õ�����(E)ģ�ͣ�������������£�

ʽ�У�![]() Ϊ�Ͻ�������ƣ�
Ϊ�Ͻ�������ƣ�![]() ��
��![]() �ֱ�
�ֱ�
Ϊa��b��ֵ������ƣ�f a(r)��f b(r)�ֱ�Ϊa��b��ֵĵ����ܶȺ�����
���о����Ӷ���ѧģ��������500��ԭ�ӵ����������н��У����õ��µ�ѹ(NPT)ϵͳ��ʹ�������Ա߽��������Ͻ���ϵ������298 K��ԥ105��ʱ�䲽������ȷ����ϵ��ֳ�ԥ�ﵽƽ��״̬��Ȼ������������8.5��1012 K/s������1 498 K�������1 498 Kʱ�ٳ�ԥ105��ʱ�䲽����ʱ�䲽��ѡ��3 fs���ڴ˹�����ѹǿ����0 Pa��
2 ���������
ͼ1��ʾΪPb8Au2��Pb9Au1������ϵ�����¹����кϽ�����������¶ȵı仯���ߡ���ͼ1�п��Կ���������õ�Pb8Au2��Pb9Au1�Ͻ���۵��¶ȷֱ�Ϊ523 K��521 K������[18]��Pb8Au2��Pb9Au1�Ͻ��ʵ���۵�ֱ�Ϊ500 K��530 K��ģ���������ֱ�Ϊ4.6%��1.7%��
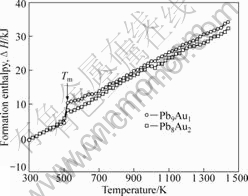
ͼ1 ��ͬ�¶��ºϽ��������
Fig.1 Formation enthalpies of alloys at different temperatures
���ݺϽ���ϵ�����ʣ����Է���ؼ�����ϵ�����¹���������ѧ���ʡ��Ͻ���ϵ���������������¶ȵĹ�ϵ���Ա�ʾ���£�
![]()
�������۵�λ��ʱ����ϵ��Һ̬���������ܵ��ڹ�̬���������ܣ�����![]() ��������۵�λ���У�
��������۵�λ���У�
![]()
����õ�Pb8Au2��Pb9Au1�Ͻ���ۻ��طֱ�Ϊ4.48 J/(mol��K)��11.04 J/(mol��K)���������ϼ���õ������ݣ����Ը���ʽ(7)�����������¶��£��Ͻ���ϵ��������[19]��
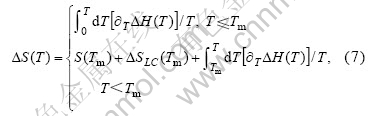
��ʽ(7)����ʽ(5)���ɼ������ͬ�¶��ºϽ���ϵ�����������ܡ�ͼ2��ʾΪ����õ���Pb8Au2��Pb9Au1���ֺϽ�����������ܡ���ͼ2�ɿ������� 1 273 Kʱ��ģ��õ���Pb8Au2��Pb9Au1�Ͻ�����������ֱܷ�Ϊ-105.21��-105.85 kJ������[18]�е�ʵ��ֵΪ-105.83��-106.04 kJ��ģ���������ֱ�Ϊ0.59%��0.18%�����Կ���ģ��ֵ������ֵ�dz� �Ǻϡ�
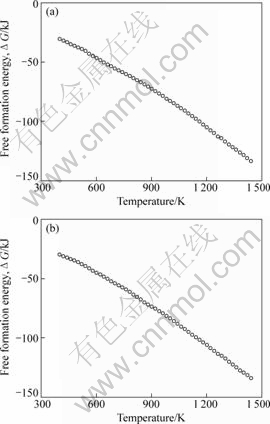
ͼ2 ��ͬ�¶��ºϽ������������
Fig.2 Free formation energies of alloys at different temperatures: (a) Pb8Au2; (b) Pb9Au1
��ԪPb-Au�Ͻ���ϵ�Ĺ�ʣ�����ܿ�����ʽ(8)����õ�
![]()
ʽ�У�![]() Ϊ�Ͻ�Ĺ�ʣ�����ܣ�
Ϊ�Ͻ�Ĺ�ʣ�����ܣ�![]() Ϊ�Ͻ�Ļ�������ܣ�
Ϊ�Ͻ�Ļ�������ܣ�![]() Ϊ�Ͻ�������������ܣ�xi��xj�ֱ�Ϊ���i��j��Ħ��������
Ϊ�Ͻ�������������ܣ�xi��xj�ֱ�Ϊ���i��j��Ħ��������
�Ͻ�Ļ�������ܿ�����ʽ(9)����õ���
![]()
ʽ�У�![]() ��
��![]() Ϊ��i��j�����ڲ�ͬ�¶��µ����������ܣ�
Ϊ��i��j�����ڲ�ͬ�¶��µ����������ܣ�![]() Ϊ�Ͻ����������ܣ�
Ϊ�Ͻ����������ܣ�![]() Ϊ�Ͻ�Ļ�������ܡ�
Ϊ�Ͻ�Ļ�������ܡ�
ͼ3��ʾΪ����õ���ͬ�¶��µ����ֺϽ��ʣ�����ܡ���ͼ3�ɿ�������1 273 Kʱ������õ���Pb8Au2��Pb9Au1���ֺϽ�Ĺ�ʣ�����ֱܷ�Ϊ-0.66��-0.99 kJ������ֵ[18]�ֱ�Ϊ-1.81��-1.05 kJ���ɼ�����ֵ������ֵ�������
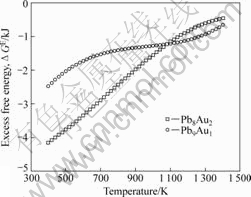
ͼ3 ��ͬ�¶��ºϽ�Ĺ�ʣ������
Fig.3 Excess free energies of alloys at different temperatures
��ͼ3�п��Կ������ֺϽ�Ĺ�ʣ�����ܾ�Ϊ��ֵ��˵���Ͻ���ԭ�Ӽ�����ýϴ���ϵΪ��ƫ����ϵ���������¶ȵ����ߣ��Ͻ�Ĺ�ʣ�������� �����¹����кϽ���ԭ��֮�������ò��Ͻ��ͣ����������ںϽ�ķ��롣
�Ͻ���ϵ��ԭ��֮�������û������ɺϽ�Ľ�������жϡ�ͼ4��ʾΪ����õ��IJ�ͬ�¶��½������ʼ��Ͻ����ܡ���ͼ4�ɿ�������1 273 Kʱ��Pb8Au2��Pb9Au1���ֺϽ�Ľ���ֱܷ�Ϊ2.07��1.87 eV����298 Kʱ��Pb��Au�����ļ�������Ϊ1.94��3.85 eV������ֵ[20]�ֱ�Ϊ2.04��3.93 eV��ģ���������Ϊ4.0%��2.0%���ɴ˿��Կ��������߷��ϽϺá�
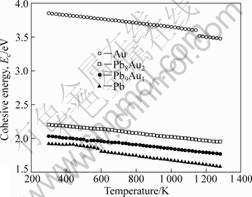
ͼ4 �ڲ�ͬ�¶��½������Ͻ�Ľ����
Fig.4 Cohesive energies of metal and alloys at different temperatures
��ͼ4�л����Կ�����1) ��ͬ�¶��£���Ǧ�Ľ����С�ڴ���Ľ���ܣ�˵��Pbԭ�ӽϽ�ԭ�Ӹ������γɹ���ԭ�ӣ�����ϱ���Ϊ��ͬ�¶���Ǧ������ѹ�Ƚ�ߣ�2) Pb-Au��Ԫ�Ͻ�Ľ�������¶ȵ����߲��Ͻ��ͣ����Ͻ���ԭ�Ӽ������ò��Ͻ��ͣ�˵���¶�Խ�ߣ�Һ̬�Ͻ����Խ���γɹ����Ľ���ԭ�ӣ�ʹ����������ѹ�������ߣ����¹��������ڽ���������������ͨ���������ѧ�����õ��Ľ����һ�£�3) Pb-Au�Ͻ����������ʱ����һ���¶��£�Pb�ӷ�����Au���ӷ�����ˣ�����ȡ�ýϺõķ���Ч�������������¶ȹ���ʱ��Au�����������Ļӷ����Ӷ���ʹPb-Au�Ͻ�ķ���Ч����
3 ����
1) ���������ֶ�ԪPb-Au�Ͻ�����������ܺ�ʣ�����ܣ�����ֵ������ֵ���ϽϺã�GEAM�ƿ��ԽϺõ�������Pb-Au�Ͻ���ϵ��
2) ���ֶ�ԪPb-Au�Ͻ�Ĺ�ʣ�����ܾ�Ϊ��ֵ��˵���Ͻ���ԭ�Ӽ�����ýϴ���ϵΪ��ƫ����ϵ��
3) �����¶ȵ����ߣ���Ԫ�Ͻ�Ĺ�ʣ����������˵�����¹����У��Ͻ���ԭ��֮�������ò��Ͻ��ͣ����������ںϽ�ķ��롣
4) ��Ԫ�Ͻ�Ľ�������¶ȵ����߲��Ͻ��ͣ�������ϵ�ڸ�ԭ��֮�����������¶ȵ����߲��Ͻ��ͣ�������һ���¶�ʱ�����ںϽ�ķ��룬���¶ȹ���ʱ������Ч������
REFERENCES[1] ������, �� ��, ���Ļ�, ��Ϊ��, ������. ��ɫ�������ұ���չ[J]. ����������ѧѧ��(������), 2004, 29: 1-4.
DAI Yong-nian, YANG Bin, MA Wen-hui, CHEN Wei-liang, DAI Jian-qing. Advances on vacuum metallurgy of nonferrous metals[J]. Journal of Kunming University of Science and Technology (Science and Technology), 2004, 29: 1-4.
[2] HAGER J P, WILKOMIRSKY R A. Galvanic cell studies using a molten oxide eletrolyte: Part ��Thermodynamic properties of the Pb-Au system[J]. Trans Met Soc AIME, 1969, 245: 2307-2312.
[3] KLEPPA O J. A thermodynamic study of liquid metallic solutions.�� The system lead�Cgold[J]. Am Chem Soc, 1949, 71(10): 3275-3280.
[4] DAW M S, BASKES M I. Embedded atom method: Derivation and application to impurities, surfaces, and other defects in metals[J]. Phys Rev B, 1984, 29(12): 6443-6453.
[5] FOILS S M, BASKES M I, DAW M S. Embedded atom method functions for the fcc metals Cu, Ag, Au, Ni, Pd, Pt and their alloys[J]. Phys Rev B, 1986, 33(12): 7983-7991.
[6] CHERNE F J, BASKES M I, HOLIAN B L. Predicted transport properties of liquid plutonium[J]. Phys Rev B, 2003, 67(9): 2104-2107.
[7] WANG L, CONG H, ZHANG Y N, BIAN X F. Medium-range order of liquid metal in the quenched state[J]. Physica B, 2005, 355: 140-146.
[8] CARO A, CARO M, LOPASSO E M, TURCHIPEA, FARKAS D. Thermodynamics of Fe-Cu alloys as described by a classic potential[J]. Journal of Nuclear Materials, 2006, 349: 317-326.
[9] HAO Xiao-gang, YU Qiu-ming, JIANG Shao-yi, SCHWARTZ D T. Molecular dynamics simulation of ion selectivity traits of nickel hexacyanoferrate thin films[J]. Trans Nonferrous Met Soc China, 2006, 16: 897-902.
[10] YANG Xi-yuan, WU Dan. Atomic simulations for surface-initiated melting of Nb(111)[J]. Trans Nonferrous Met Soc China, 2009, 19: 210-214.
[11] ��, ������. Ag��ָ�������гЧӦ�ķ��Ӷ���ѧģ��[J].�й���ɫ����ѧ��, 2005, 15(11): 1859-1863.
YANG Jian-yu, HU Wang-yu. Molecular dynamics simulation of an harmonic effects for low Miller index surfaces of Ag[J]. The Chinese Journal of Nonferrous Metals, 2005, 15(11): 1859-1863.
[12] ��־��, �� ��, ������, ������, ����, ��־��. Ni-Al�Ͻ����̹��̵ķ��Ӷ���ѧģ��[J]. �й���ɫ����ѧ��, 2009, 19(8): 1409-1415.
ZHU Zhi-xiong, ZHANG Hong, LIU Chao-feng, QI Wei-hong, YI Dan-qing, LI Zhi-cheng. Molecular dynamics simulation for solidification process of Ni-Al alloys[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(8): 1409-1415.
[13] LIU J, ZHAO J Z, HU Z Q. MD study of the glass transition in binary liquid metals: Ni6Cu4 and Ag6Cu4[J]. Intermetallics, 2007, 15: 1361-1366.
[14] QI L, ZHANG H F, HU Z Q. Molecular dynamic simulation of glass formation in binary liquid metal: Cu-Ag using EAM[J]. Intermetallics, 2004, 12: 1191-1195.
[15] WADLEY H N G, ZHOU X W, JOHNSON R A, NEUROCK M. Mechanisms, model and methods of vapor deposition[J]. Progress in Material Science, 2001, 46: 329-377.
[16] ZHOU X W, WADLEY H N G, JOHNSON R A, LARSON D J, TABAT N, CERZO A. Atomic scale structure of sputtered metal multilayers[J]. Acta Mater, 2001, 49: 4005-4015.
[17] JOHNSON R A. Analytic nearest-neighbor modle for fcc metals[J]. Phys Rev B, 1988, 37: 3924-3930.
[18] HULTGREN R, DESSAI P D, HAWKINS D T. Selected values of the thermodynamic properties of binary alloy[M]. Ohio: ASM Metal Park, 1973.
[19] TEICHER H. Melting transition in molecular-dynamics simulations of the Ni0.5Zr0.5 intermetallic compound[J]. Phys Rev B, 1999, 59(13): 8473-8479.
[20] SMITH C J. Metals reference book[M]. London: Butterworrd, 1976.
(�༭ ����)
�ո����ڣ�2009-05-21�������ڣ�2009-09-15
ͨ�����ߣ��� ���ڣ���ʿ���绰��0871-5161583��E-mail��kgyb2005@126.com
